
Abstract
Intellectual disability (ID) is a heterogeneous condition affecting 2–3% of the population, often associated with multiple congenital anomalies (MCA). The genetic cause remains largely unexplained for most cases. To investigate the causes of ID/MCA of unknown etiology in the Japanese population, 645 subjects have been recruited for the screening of pathogenic copy-number variants (CNVs). Two screenings using bacterial artificial chromosome (BAC) arrays were previously performed, which identified pathogenic CNVs in 133 cases (20.6%; Hayashi et al., J. Hum. Genet., 2011). Here, we present the findings of the third screening using a single-nucleotide polymorphism (SNP) array, performed in 450 negative cases from our previous report. Pathogenic CNVs were found in 22 subjects (4.9%), in which 19 CNVs were located in regions where clinical significance had been previously established. Among the 22 cases, we identified PPFIA2 as a novel candidate gene for ID. Analysis of copy-neutral loss of heterozygosity (CNLOH) detected one case in which the CNLOH regions seem to be significant. The SNP array detected a modest fraction of small causative CNVs, which is explained by the fact that the majority of causative CNVs have larger sizes, and those had been mostly identified in the two previous screenings.
Similar content being viewed by others


Introduction
Intellectual disability (ID), frequently associated with multiple congenital anomalies (MCA), is defined as a cluster of syndromes and disorders characterized by low intelligence and associated limitations in adaptive behavior,1 affecting ~2–3% of the population.2 It has been estimated that 25–50% are attributed to genetic causes.3 Nevertheless, the etiology remains largely unknown for a significant proportion of the cases.
Chromosomal imbalances and copy-number variations (CNVs) are frequent causes of many developmental and genetic disorders, including ID/MCA. Such changes, in the form of submicroscopic deletions or duplications, can lead to a disease (or increase its susceptibility) through an abnormal dosage of one or more genes located within the rearranged segments, disruption of one or more genes or by the unmasking of a recessive allele. Over the past years, the application of chromosomal microarray analysis (CMA) has revolutionized the diagnosis of children presenting with ID/MCA and several other disorders, as the use of CMA in subjects with an apparently normal karyotype has increased the diagnostic yield by an additional 12% on average.4
Aiming at identifying the etiology behind those conditions in the Japanese population, we have launched a project that started in 2005 (Japanese Array Consortium), with the recruitment of 645 subjects presenting with clinically uncharacterized ID/MCA. Results from a two-stage screening using a targeted and whole-genome bacterial artificial chromosome (BAC) arrays are described in Hayashi et al.5 Here, we present our findings in the third screening performed with a single-nucleotide polymorphism (SNP) array, applied to 450 subjects in whom pathogenic CNVs were not detected in the two previous screenings. The use of SNP arrays is advantageous because, besides a higher resolution over BAC arrays, the SNP genotyping provides information about uniparental disomy (UPD) and parental consanguinity, through the detection of copy-neutral loss of heterozygosity (CNLOH).
Materials and methods
Subjects
We constructed a consortium composed of 23 institutions in Japan, with the recruitment of 645 Japanese subjects over 10 years of project. All patients were examined by specialists in Medical Genetics and were referred to cytogenetic testing owing to the presence of various findings such as unexplained ID, developmental delay and dysmorphic features. The majority consisted of sporadic cases, and all patients have had a previous result of normal karyotype. Signed informed consents were obtained for all the subjects, along with the approval by the local ethics committee of all institutions involved in this project. Genomic DNA was extracted from peripheral blood following standard procedures, and lymphoblastoid cell lines establishment by Epstein–Barr virus immortalization followed the procedures as described previously.6
Before the third screening, all subjects had been submitted to a two-stage screening using two in-house BAC arrays. Initially, 536 patients were screened, and pathogenic CNVs were found in 18.7% of the cases (100/536).5 Subsequently, 109 patients were further recruited and screened by the two BAC arrays, with the identification of an additional number of 33 pathogenic CNVs (Supplementary Tables 1 and 2). After the second screening, all the subjects that had no CNVs identified (432 cases) or were detected with only benign CNVs (18 cases) composed our final cohort for the third screening, consisting of 450 subjects. A schematic showing the number of patients in each screening is depicted in Supplementary Figure 1.
The control cohort for the third screening consisted of 100 parent–child trios of healthy Brazilian individuals of full Japanese ancestry, and these data are also available in the MCG CNV Database (http://www.cghtmd.jp/CNVDatabase/).
SNP array analysis
SNP genotyping was performed using the HumanOmniExpress-12 v1.0 DNA Analysis BeadChip (Illumina, San Diego, CA, USA), following the manufacturer’s instructions. Slides were scanned on an iScan Microarray Scanner (Illumina), and CNVs were called by KaryoStudio v1.4 (Illumina) under the cnvPartition v3.0.7 plug-in algorithm. All CNVs were mapped within the GRCh37/hg19 human genome assembly. For gains, we used the threshold of >50 kb that is commonly used in SNP array analysis,7, 8 and a more optimized threshold of >10 kb for losses, to avoid missing smaller causative variants. For CNLOH, the cut-off length was set at 3 Mb, following the minimal threshold defined for clinical analyses.9
Data interpretation
The CNV calls were evaluated on the basis of the following criteria: gene content (including function and expression), previous reports in the literature, presence of genes cataloged in the Online Mendelian Inheritance in Man (OMIM), number of overlapping CNVs in the Database of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources (DECIPHER) and in the Database of Genomic Variants (DGV). Parental analysis for cases in which samples were available also helped in the assessment of CNV pathogenicity.
Confirmation of the results by a second independent assay
For subjects in whom CNVs of clinical relevance or unknown significance were detected, validation of the SNP arrays results were performed with either fluorescence in situ hybridization (FISH) or quantitative real-time PCR (qPCR) in samples of the patients and the parents, whenever possible. Fluorescence in situ hybridization was performed following standard protocols and using BAC clones located in the region of interest.10 qPCR was performed with the KAPA SYBR FAST qPCR Master Mix (KAPA Biosystems, Wilmington, MA, USA) on a 7500 Real-Time PCR System (Applied Biosystems, Carlsbad, CA, USA). Data analysis was carried out using the comparative threshold cycle method.11 The qPCR primer sequences are available upon request.
Methylation-specific PCR
DNA samples were subjected to bisulfite conversion using the EZ DNA Methylation kit (Zymo Research, Irvine, CA, USA). Methylation-specific PCR in NNAT locus in Patient 38 was performed using the following primers: 5′-GATTGGCGGTTTAAAAGGGATTC-3′ and 5′-CTATACGACTAAATCACCGAACG-3′ (methylated alleles); 5′-GATTGGTGGTTTAAAAGGGATTT-3′ and 5′-CTCCCCCAAACCCTAATAAATCA-3′ (unmethylated alleles). Protocol and PCR cycling program were adapted from Kubota et al.,12 with minor modifications. The products were separated by electrophoresis on 3% agarose gel.
Complementary DNA synthesis
Total RNA was isolated from Epstein–Barr virus-transformed lymphoblastoid cell lines using TRIsure (Bioline, London, UK) and treated with DNase I (Takara Bio, Shiga, Japan). Complementary DNA synthesis was obtained with the PrimeScript II first-strand complementary DNA Synthesis kit (Takara Bio) using oligo dT primers.
Microsatellite genotyping
For confirmation of UPD in Patient 38, 13 microsatellite markers along chromosome 20 were genotyped (D20S603, D20S846, D20S470, D20S875, D20S54, D20S471, D20S200, D20S843, D20S195, D20S884, D20S107, D20S75 and D20S840). We followed the method developed by Schuelke.13 The PCR products were separated by capillary electrophoresis on an ABI 3730xl DNA Analyzer (Applied Biosystems) with the GeneScan 500 LIZ size standard (Applied Biosystems). Fragment analysis was performed with the GeneMapper software (Applied Biosystems).
Results
CNV analysis
CNVs with potential clinical relevance
The screening of 450 patients with undiagnosed ID/MCA by SNP arrays detected rare 23 CNVs with potential clinical relevance in 22 cases (Table 1). One case (Patient 16) was found with two CNVs. No similar CNVs were found in the control cohort. Parental samples were available for inheritance assessment in eight cases, in which four occurred de novo. The duplication of Patient 19 was inherited from the unaffected father, who has the same rearrangement in mosaic state.
Briefly, we classified the clinically relevant CNVs into the following categories: single-gene deletions, copy-number gains, CNVs overlapping recently established syndromes, recurrent CNVs in known susceptibility regions and large rearrangements over 10 Mb. Two cases had been simultaneously investigated by other related groups and had the same results achieved, and those were already reported: a SMARCA2 deletion in Patient 614 and a CREBBP deletion in Patient 18.15
Single-gene deletions
Single-gene deletions were identified in heterozygous state in three cases, all being intragenic. The most representative case is Patient 9, detected with a de novo 186-kb deletion at 12q21.31 involving PPFIA2 (Figure 1), which might be a novel gene associated with ID. The patient is a 6-year-old male with mild ID, intrauterine growth retardation and minor anomalies. The deletion is in-frame and encompasses the exons 5, 6 and 7 of PPFIA2 (NM_003625), a region in which benign variants have not been described so far in the Database of Genomic Variants (Figure 1b). PPFIA2 is highly expressed solely in brain16 and binds directly to the known X-linked ID gene CASK in the MALS-CASK-Mint-1 complex.17 This interaction is unique to vertebrates and likely to regulate higher-order brain functions in mammals.18 Although CASK mutations or heterozygous deletions in females are known to cause ID and microcephaly with pontine and cerebellar hypoplasia19, 20 (OMIM #300749), brain magnetic resonance imaging of the patient was normal.

Representative deletion detected in the third screening. (a) SNP array profile showing the 186-kb deletion at 12q21.31 found in Patient 9, depicted by a box. (b) Localization of the deletion (represented by a box), in which three exons of the five longer isoforms of PPFIA2 are deleted, with no shift in the reading frame. The region neighboring the three exons has no benign variants described in the Database of Genomic Variants (track shown in dense mode in the UCSC genome browser). (c) FISH performed with BAC clones at 12q12 (RP11-525K7, red) and at 12q21.31 (RP11-259P16, green) revealed that the deletion in Patient 9 is de novo (arrow). BAC, bacterial artificial chromosome; CNV, copy-number variant; FISH, fluorescence in situ hybridization; SNP, single-nucleotide polymorphism. A full color version of this figure is available at the Journal of Human Genetics journal online.
Copy-number gains
Copy-number gains, mostly duplications, accounted for 31.8% (7/22) of the cases with clinically relevant CNVs. The two most representative cases are described below.
Patient 17, a 4-year-old female with severe ID, short stature, microcephaly and Chiari malformation, was detected with a 71-kb gain encompassing IGF1R at 15q26.3 (Figures 2a and b). Fluorescence in situ hybridization revealed that the amplified segments are in tandem (Supplementary Figure 2a). Although the KaryoStudio software called this CNV with a copy-number value of three, qPCR indicated the presence of four copies of IGF1R, thereby meaning that the rearrangement might be a triplication (Figure 2c). In addition, qRT-PCR performed on complementary DNA from lymphoblastoid cell line revealed downregulation of IGF1R (Figure 2d). This suggests the triplication may have a more complex structure that disrupted one allele, thus leading to haploinsufficiency of IGF1R. As IGF1R heterozygous aberrations have been long known to cause intrauterine growth retardation and short stature,21, 22 the IGF1R haploinsufficiency is likely responsible for the short stature of Patient 17.

Representative copy-number gains found in the third screening. Patient 17: (a) SNP array profile of the 71-kb gain at 15q26.3 in Patient 17, highlighted by a box. (b) Position of the gain (represented by a box), encompassing part of IGF1R. (c) qPCR of copy number suggested the presence of four copies of IGF1R in the Patient 17, implying that the rearrangement is actually a triplication. (d) qRT-PCR performed in LCL suggested downregulation of IGF1R in Patient 17. Patient 19: (e) SNP array profile showing a 333-kb duplication at 16p13.2 in Patient 19, represented by a box. (f) Schematic representation of the duplication. USP7 and C16orf72 are shown relative to the reference genome (above) and the duplication (below). The dashed-line rectangles depict the duplicated region, and arrowheads represent the primers used for breakpoint mapping. (g) Duplication-specific PCR (using the primers shown in f) detected a faint product in the father, suggesting low-level gonosomal mosaicism. (h) Partial electropherograms showing the breakpoint junction of the duplication, confirming that the father also has the duplication, although in mosaic state. The duplication has an insertion of 6 bp in the breakpoint region. LCL, lymphoblastoid cell line; qPCR, quantitative PCR; qRT-PCR, quantitative reverse transcribed PCR; SNP, single-nucleotide polymorphism. A full color version of this figure is available at the Journal of Human Genetics journal online.
Patient 19 is a 5-year-old female with severe ID and autistic tendency. We detected a 333-kb duplication at 16p13.2 encompassing USP7 and C16orf72 (Figures 2e and f). Although qPCR suggested that the duplication was de novo (Supplementary Figure 2b), a duplication-specific PCR showed a faint band corresponding to the amplification of the breakpoint junction in the father (Figure 2g). The sequencing of the faint band revealed the existence of the identical breakpoint junction seen in the proband, pointing to gonosomal mosaicism of this rearrangement in the father (Figure 2h). The 16p13.2 locus has been previously identified as a novel autism spectrum disorder locus, where three duplications (two de novo and one inherited) were detected in autism spectrum disorder patients.23 As the minimum common interval also comprised USP7 and C16orf72, the duplication in Patient 19 might explain her tendency to autism.
CNVs overlapping recently established syndromes
Six cases overlapped with recently established syndromes: Patient 1 (Chromosome 1q43-q44 deletion syndrome—OMIM #612337), Patient 3 (2p14p15 microdeletion syndrome24), Patient 5 (Chromosome 4q21 deletion syndrome—OMIM #613509), Patient 7 (10q11.21q11.23 deletion/duplication syndromes25), Patient 21 (Chromosome 16p11.2 duplication syndrome—OMIM #614671) and Patient 22 (Sotos syndrome 2—OMIM #614753).
Recurrent CNVs in known susceptibility regions
Recurrent chromosomal microdeletions and microduplications predisposing to neurodevelopmental disorders, in which frequent ‘hotspots’ include the 15q11.2, 15q13.3, 16p11.2 and 22q11.2 loci,26, 27 corresponded to 31.8% (7/22) of the patients with clinically relevant CNVs in our study. Notably, we observed three deletions (Patients 10, 11 and 12) and two duplications (Patients 13 and 14) in the 15q11.2 region delimited by breakpoints (BP) 1 and 2. Collectively, the frequency of 0.77% (5/645) of BP1–BP2 rearrangements in our cohort is consistent with those found in previous studies (0.86% in Burnside et al.28 and 0.8% in Vanlerberghe et al.29). Although BP1–BP2 rearrangements in healthy individuals have been observed with a prevalence of 0.25% in previous studies,30 we did not detect any in our control group.
Although deletions involving the 15q13.3 and 16p13.11 loci are regarded as pathogenic, gains in these regions have unknown clinical significance. Interestingly, we detected a girl (Patient 16) concomitantly with a 491-kb duplication at 15q13.3 and a 3-Mb duplication at 16p13.11p12.3, respectively inherited from the father and the mother. This might indicate that both duplications could have contributed in an additive or epistatic manner, compatible with the two-hit model.31
Large rearrangements over 10 Mb
Two very large rearrangements were detected by SNP array: an 11.7-Mb duplication at 11q23.3q24.3 (Patient 8) and an 18.5-Mb deletion at 3q13.12q21.3 (Patient 4). Although these CNVs had been detected in the second BAC array screening, SNP array was performed to discriminate several other CNVs from artifacts. The parental origin was not investigated. However, the pathogenicity of these CNVs is certainly clear because of their very large sizes.
Variants of uncertain clinical significance
All CNVs with insufficient evidence to be determined as either pathogenic or benign were classified as variants of uncertain clinical significance. Among the 450 cases, 15 fell into this category (Supplementary Table 3). This category involves frequent findings in clinical cytogenetics screenings, such as 16p13.11 duplications (Patient 32), deletions/duplications involving ASTN2 (Patients 26 and 27), Xp22.31 microduplications (Patients 34 and 35) and, most notoriously, CHRNA7 duplications at 15q13.3 (Patients 29, 30 and 31). The remaining cases either refer to CNVs that might be clinically relevant, but lack of full parental investigation did not allow their definitive classification as such (Patients 23, 24, 25, 33 and 36), or cases where uncertainty still exists even after inheritance determination (Patients 28 and 37).
CNLOH analysis
When analyzing CNLOH data provided by the SNP genotyping, two criteria that may imply clinical importance must be distinguished: an isolated CNLOH, usually longer than 20 Mb, likely representative of a UPD event,32 and an excess of homozygous regions spread over the genome, representing regions of identity by descent. Five cases fulfilling the criteria above were identified in the screening (Table 2).
Isolated CNLOH
We identified only one case with a single CNLOH over 20 Mb in Patient 38, a 13-year-old male with mild ID, developmental delay and minor anomalies (Table 2). The CNLOH is almost 24 Mb in size and encompasses the centromere of chromosome 20 (20p12.1q11.23; Figure 3a). This region contains two genes subjected to genomic imprinting, BLCAP and NNAT. The SNP array analysis in the parent–child trio and microsatellite genotyping using 13 markers along chromosome 20 did not reveal any informative markers for confirmation of UPD. We tested another approach by investigating the methylation status of NNAT, because, like many imprinted genes, NNAT shows a differential methylation of the CpG islands. Methylation-specific PCR for NNAT locus suggested the presence of biparental alleles in the proband (Figure 3b). This result is actually not unusual, considering that Papenhausen et al.32 found out that, of 46 cases, UPD was not validated in 16. Similarly, they observed that the false positive cases had a greater frequency of centromeric involvement. Nonetheless, the possibility remains that the phenotype of Patient 38 might be explained by a recessive mutation located in the CNLOH.

CNLOH analysis in the third screening. (a) SNP array profile of chromosome 20 in Patient 38, showing a large region of homozygous markers (highlighted by a rectangle), including the centromere. (b) MS-PCR of NNAT locus revealed that Patient 38 has both methylated and unmethylated alleles, suggesting biparentality (M: methylated, U: unmethylated). (c) CNLOH pattern of Patient 39, showing the distribution of CNLOH >3 Mb in seven chromosomes, as visualized in the Illumina KaryoStudio v1.4 software. The CNLOH regions are represented by blue bars, left to each chromosome ideogram. Sex chromosomes are excluded in CNLOH modeling. (d) CNLOH in Patient 40, showing large homozygous regions especially in chromosomes 2 and 3, as visualized in the Illumina KaryoStudio v1.4 software. The CNLOH regions are represented by blue bars, left to each chromosome ideogram. CNLOH, copy-neutral loss of heterozygosity; MS-PCR, methylation-specific PCR; SNP, single-nucleotide polymorphism. A full color version of this figure is available at the Journal of Human Genetics journal online.
Excessive homozygosity throughout the genome
Among the cases with CNLOH in more than two chromosomes, excessive homozygosity was detected in four patients (Table 2). Two cases (41 and 42) that were previously known to be offspring of first-cousin marriages were detected with an autozygosity of 4.98 and 6.34%, respectively. On the other hand, the parents of Patients 39 and 40 were reportedly unrelated. CNV analysis did not reveal any imbalances that could be of clinical relevance; hence the clinical presentation of each patient is presumably caused by a recessive point mutation. Except for Patient 39, parental samples were not available for further investigations.
A total of 40-Mb CNLOH (autozygosity of 1.4%, Figure 3c) was detected in Patient 39, a male proband (IV-9) from a familial case of joint hyperextensibility, where the father (III-3), an aunt (III-2) and great grandfather (I-1) from the paternal side were also affected (Supplementary Figure 3). In addition, the proband presented a few features resembling Aarskog–Scott syndrome (OMIM #305400), like short stature, brachydactyly, shawl scrotum and hyperactivity. An investigation of FGD1, the causative gene of Aarskog–Scott syndrome, had been previously negative. Details of this family will be described elsewhere (Uehara et al., manuscript in preparation).
Patient 40, a girl with severe ID, short stature, microcephaly, tendency to hirsutism, no speech, stereotypic hands movements, and two-to-three toe syndactyly, had very large CNLOH regions detected in chromosomes 2 and 3, besides a 3.4-Mb CNLOH in chromosome 10 (Figure 3d). Interestingly, the CNLOH in chromosome 2 include regions that have been associated with syndromes whose features partially match those seen in the patient: the Filippi syndrome (OMIM #272440), caused by recessive mutations in the CKAP2L gene at 2q13; and the Chromosome 2q23.1 deletion syndrome (OMIM #156200), caused by heterozygous mutations or disruption of MBD5. We attempted to sequence CKAP2L and MBD5, but novel variants were not found in both genes. Nonetheless, the possibility of a recessive mutation in other genes is not discarded.
In addition, large CNLOH in two chromosomes were also found in an individual from our control cohort: a 35-Mb and a 33-Mb segments in chromosomes 2 and 8, respectively (data not shown). This illustrates how very large homozygous regions can be found also in healthy individuals, with no apparent clinical consequences.
Point mutations later identified in subjects negative for relevant CNVs
Among the 450 patients submitted to SNP array, some patients with no relevant CNVs were later clinically diagnosed based on their late-onset phenotypes. The respective causative genes were sequenced and point mutations were identified in 21 subjects (Supplementary Table 4).
Discussion
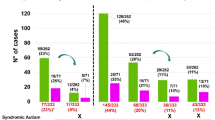
We have performed three screenings utilizing two types of BAC array and a SNP array in 645 Japanese subjects presenting with variable phenotypic expressivity and severity of undiagnosed ID/MCA. Overall, our three-stage screening allowed the identification of pathogenic CNVs in 24% of the cases (155/645). More specifically, this category was identified in 22 subjects in the third screening by SNP array, representing 3.4% of our total cohort (22/645). Figure 4 summarizes the results obtained in the three screenings.

Summary of the results obtained in each screening. Amid the first to second screenings, 51 cases were canceled and not submitted to subsequent analyses. BAC, bacterial artificial chromosome; CNLOH, copy-neutral loss of heterozygosity; CNV, copy-number variant; SNP, single-nucleotide polymorphism; VOUS, variant of uncertain clinical significance.
Collectively, large CNVs (>1 Mb) represent 81.3% of our positive cases (126/155), of which 117 were detected in the first and second screenings. This is in agreement with the observation that most of the causative CNVs have large sizes,33, 34 provided that larger CNVs frequently contain more genes, thus having a higher chance of playing a role in the alteration of physiological functions.35 Because we adopted a strategy of performing three screenings, starting from relatively low to a high-resolution array, the low number of causative CNVs identified in the third screening is not unexpected.
Among the 22 subjects with clinically relevant CNVs identified in the third screening, a rare de novo PPFIA2 deletion (Patient 9) is the only case to reveal a potential candidate gene for ID. So far, PPFIA2 has only been suggested as a candidate gene to high-grade myopia, in which an association study identified an intronic SNP that showed a significant association with that ocular disease.36 PPFIA2, or Liprin-α2, belongs to the liprin family of LAR transmembrane protein-tyrosine phosphatases that organize the presynaptic active zone and regulate neurotransmitter release.17 The fact that PPFIA2 is only expressed in brain and interacts with the MALS–CASK–Mint1 complex, through the direct binding to the known X-linked ID CASK, suggest a plausible pathogenic role of PPFIA2 haploinsufficiency in the phenotype of Patient 9.
The CNLOH analysis revealed two patients (39 and 40) with relative high percentages of homozygosity, but kinship relationship was not mentioned for their respective parents. In Patient 40, the large CNLOH regions might represent non-disjunction events rather than identity by descent. As parental samples could not be examined, it remains unclear whether those regions have a clinical significance, provided that we also found a control individual with very large CNLOH in two chromosomes. Furthermore, long stretches of homozygous regions are not unusual even in outbred populations and are thought to reflect the presence of ancestral haplotypes that remain intact owing to locally low rates of recombination.37 On the other hand, the CNLOH found in Patient 39 were distributed in seven chromosomes, with an autozygosity of 1.4%. As an autozygosity of ~1.5% is to be expected in the offspring of a second-cousin marriage, the relative high number of homozygous regions in Patient 39 might be owing to the fact that his family is from a relatively isolated population in Japan, the southernmost island of Okinawa, where there is less genetic variability than in mainland Japan.38
After the third screening, 408 subjects had no relevant CNVs identified (Figure 4). Excluding the 21 cases in which point mutations were detected afterwards (Supplementary Table 4), this number drops to 387. This means that at least 60% (387/645) of the total cohort remains with unknown etiology. These cases might be explained by point mutations, small deletions and insertions, epigenetic alterations, and structural aberrations not detected by microarrays, such as balanced translocations and inversions. Moreover, it is possible that the SNP array did not detect microimbalances in mosaic state that are present in tissues other than peripheral blood. Finally, it should be noted that SNP arrays have their resolution limited by the SNP distribution and signal to background,39 with a risk of leaving causative CNVs undetected.
It must be mentioned that we detected, in the third screening, a few rare CNVs encompassing genes whose function was either unknown or not clearly correlated with the phenotypic features of a given patient. The unavailability of parental samples prevented us from identifying possible de novo variants among those CNVs. Thus, we classified all of them as benign. If de novo, we would have proceeded to additional investigations that not only might have allowed the identification of novel genes related to ID, but also provided clues upon their function. It is evident how a precise clinical interpretation requires that the analysis be extended to parents as much as possible.
Our findings are consistent with previous studies that found pathogenic CNVs in 15–20% of the cases studied.4 As the etiology of 60% of the cases continue to be unexplained, it would be desirable to apply other approaches for elucidating the cause in those subjects. With the advances provided by the application of high-throughput sequencing technologies, patients suffering from undiagnosed conditions have been targeted by initiatives such as the Undiagnosed Diseases Network of the United States National Institutes of Health, and the Deciphering Developmental Disorders Study from the Health Innovation Challenge Fund and the Wellcome Trust Sanger Institute, in the United Kingdom. By combining whole-exome sequencing, exome-focused array comparative genomic hybridization and SNP genotyping, a report by the Deciphering Developmental Disorders project was able to increase by 10% the proportion of children that could be diagnosed, besides the identification of 12 novel genes associated with developmental disorders.40 As high-throughput sequencing technologies are gradually becoming more accessible, the full integration of such tools in future investigations will be truly valuable for increasing the diagnostic yield as well, under the adoption of suitable study designs.
Web resources
The web resources used for this research are DECIPHER (Database of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources): http://decipher.sanger.ac.uk/; DGV (Database of Genomic Variants): http://dgv.tcag.ca/dgv/app/home; and OMIM (Online Mendelian Inheritance in Man): http://www.ncbi.nlm.nih.gov/omim.
References
Salvador-Carulla, L., Reed, G. M., Vaez-Azizi, L. M., Cooper, S. A., Martinez-Leal, R., Bertelli, M. et al. Intellectual developmental disorders: towards a new name, definition and framework for "mental retardation/intellectual disability" in ICD-11. World Psychiatry 10, 175–180 (2011).
de Vries, B. B ., Pfundt, R., Leisink, M., Koolen, D. A., Vissers, L. E., Janssen, I. M. et al. Diagnostic genome profiling in mental retardation. Am. J. Hum. Genet. 77, 606–616 (2005).
Kaufman, L., Ayub, M. & Vincent, J. B. The genetic basis of non-syndromic intellectual disability: a review. J. Neurodev. Disord. 2, 182–209 (2010).
Miller, D. T., Adam, M. P., Aradhya, S., Biesecker, L. G., Brothman, A. R., Carter, N. P. et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet. 86, 749–764 (2010).
Hayashi, S., Imoto, I., Aizu, Y., Okamoto, N., Mizuno, S., Kurosawa, K. et al. Clinical application of array-based comparative genomic hybridization by two-stage screening for 536 patients with mental retardation and multiple congenital anomalies. J. Hum. Genet. 56, 110–124 (2011).
Saito-Ohara, F., Fukuda, Y., Ito, M., Agarwala, K. L., Hayashi, M., Matsuo, M. et al. The Xq22 inversion breakpoint interrupted a novel Ras-like GTPase gene in a patient with Duchenne muscular dystrophy and profound mental retardation. Am. J. Hum. Genet. 71, 637–645 (2002).
Männik, K., Parkel, S., Palta, P., Zilina, O., Puusepp, H., Esko, T. et al. A parallel SNP array study of genomic aberrations associated with mental retardation in patients and general population in Estonia. Eur. J. Med. Genet. 54, 136–143 (2011).
D'Amours, G., Langlois, M., Mathonnet, G., Fetni, R., Nizard, S., Srour, M. et al. SNP arrays: comparing diagnostic yields for four platforms in children with developmental delay. BMC Med. Genomics 7, 70 (2014).
Kearney, H. M., Kearney, J. B. & Conlin, L. K. Diagnostic implications of excessive homozygosity detected by SNP-based microarrays: consanguinity, uniparental disomy, and recessive single-gene mutations. Clin. Lab. Med. 4, 595–613 (2011).
Hayashi, S., Kurosawa, K., Imoto, I., Mizutani, S. & Inazawa, J. Detection of cryptic chromosome aberrations in a patient with a balanced t(1;9)(p34.2;p24) by array-based comparative genomic hybridization. Am. J. Med. Genet. A 139, 32–36 (2005).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 4, 402–408 (2011).
Kubota, T., Das, S., Christian, S. L., Baylin, S. B., Herman, J. G. & Ledbetter, D. H. Methylation-specific PCR simplifies imprinting analysis. Nat. Genet. 16, 16–17 (1997).
Schuelke, M. An economic method for the fluorescent labeling of PCR fragments. Nat. Biotechnol. 18, 233–234 (2000).
Tsurusaki, Y., Okamoto, N., Ohashi, H., Kosho, T., Imai, Y., Hibi-Ko, Y. et al. Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome. Nat. Genet. 44, 376–378 (2012).
Kosaki, R., Fujita, H., Takada, H., Okada, M., Torii, C. & Kosaki, K. Monozygotic twins of Rubinstein-Taybi syndrome discordant for glaucoma. Am. J. Med. Genet. A 155A, 1189–1191 (2011).
Serra-Pagès, C., Medley, Q. G., Tang, M., Hart, A. & Streuli, M. Liprins, a family of LAR transmembrane protein-tyrosine phosphatase-interacting proteins. J. Biol. Chem. 273, 15611–15620 (1998).
Olsen, O., Moore, K. A., Fukata, M., Kazuta, T., Trinidad, J. C., Kauer, F. W. et al. Neurotransmitter release regulated by a MALS-liprin-alpha presynaptic complex. J. Cell Biol. 170, 1127–1134 (2005).
Wei, Z., Zheng, S., Spangler, S. A., Yu, C., Hoogenraad, C. C. & Zhang, M. Liprin-mediated large signaling complex organization revealed by the liprin-α/CASK and liprin-α/liprin-β complex structures. Mol. Cell 43, 586–598 (2011).
Hayashi, S., Mizuno, S., Migita, O., Okuyama, T., Makita, Y., Hata, A. et al. The CASK gene harbored in a deletion detected by array-CGH as a potential candidate for a gene causative of X-linked dominant mental retardation. Am. J. Med. Genet. A 146A, 2145–2151 (2008).
Najm, J., Horn, D., Wimplinger, I., Golden, J. A., Chizhikov, V. V., Sudi, J. et al. Mutations of CASK cause an X-linked brain malformation phenotype with microcephaly and hypoplasia of the brainstem and cerebellum. Nat. Genet. 40, 1065–1067 (2008).
Abuzzahab, M. J., Schneider, A., Goddard, A., Grigorescu, F., Lautier, C., Keller, E. et al. IGF-I receptor mutations resulting in intrauterine and postnatal growth retardation. N. Engl. J. Med. 349, 2211–2222 (2003).
Kawashima, Y., Kanzaki, S., Yang, F., Kinoshita, T., Hanaki, K., Nagaishi, J. et al. Mutation at cleavage site of insulin-like growth factor receptor in a short-stature child born with intrauterine growth retardation. J. Clin. Endocrinol. Metab. 90, 4679–4687 (2005).
Sanders, S. J., Ercan-Sencicek, A. G., Hus, V., Luo, R., Murtha, M. T., Moreno-De-Luca, D. et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron 70, 863–885 (2011).
Wohlleber, E., Kirchhoff, M., Zink, A. M., Kreiss-Nachtsheim, M., Küchler, A., Jepsen, B. et al. Clinical and molecular characterization of two patients with overlapping de novo microdeletions in 2p14-p15 and mild mental retardation. Eur. J. Med. Genet. 54, 67–72 (2011).
Stankiewicz, P., Kulkarni, S., Dharmadhikari, A. V., Sampath, S., Bhatt, S. S., Shaikh, T. H. et al. Recurrent deletions and reciprocal duplications of 10q11.21q11.23 including CHAT and SLC18A3 are likely mediated by complex low-copy repeats. Hum. Mutat. 33, 165–179 (2012).
Dittwald, P., Gambin, T., Szafranski, P., Li, J., Amato, S., Divon, M. Y. et al. NAHR-mediated copy-number variants in a clinical population: mechanistic insights into both genomic disorders and Mendelizing traits. Genome Res. 23, 1395–1409 (2013).
Watson, C. T., Marques-Bonet, T., Sharp, A. J. & Mefford, H. C. The genetics of microdeletion and microduplication syndromes: an update. Annu. Rev. Genomics Hum. Genet. 15, 215–244 (2014).
Burnside, R. D., Pasion, R., Mikhail, F. M., Carroll, A. J., Robin, N. H., Youngs, E. L. et al. Microdeletion/microduplication of proximal 15q11.2 between BP1 and BP2: a susceptibility region for neurological dysfunction including developmental and language delay. Hum. Genet. 4, 517–528 (2011).
Vanlerberghe, C., Petit, F., Malan, V., Vincent-Delorme, C., Bouquillon, S., Boute, O. et al. 15q11.2 microdeletion (BP1-BP2) and developmental delay, behaviour issues, epilepsy and congenital heart disease: a series of 52 patients. Eur. J. Med. Genet. 58, 140–147 (2015).
Cox, D. M. & Butler, M. G. The 15q11.2 BP1-BP2 microdeletion syndrome: a review. Int. J. Mol. Sci 16, 4068–4082 (2015).
Girirajan, S., Rosenfeld, J. A., Cooper, G. M., Antonacci, F., Siswara, P., Itsara, A. et al. A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat. Genet. 42, 203–209 (2010).
Papenhausen, P., Schwartz, S., Risheg, H., Keitges, E., Gadi, I., Burnside, R. D. et al. UPD detection using homozygosity profiling with a SNP genotyping microarray. Am. J. Med. Genet. A 155A, 757–768 (2011).
Lee, C., Iafrate, A. J. & Brothman, A. R. Copy number variations and clinical cytogenetic diagnosis of constitutional disorders. Nat. Genet. 39, S48–S54 (2007).
Girirajan, S., Brkanac, Z., Coe, B. P., Baker, C., Vives, L., Vu, T. H. et al. Relative burden of large CNVs on a range of neurodevelopmental phenotypes. PLoS Genet. 7, e1002334 (2011).
Vermeesch, J. R. & de Ravel, T. J. L. in Genetics of Mental Retardation (ed Knight, S. J. L.) 114–125 (Karger, Basel, Switzerland, 2010).
Hawthorne, F., Feng, S., Metlapally, R., Li, Y. J., Tran-Viet, K. N., Guggenheim, J. A. et al. Association mapping of the high-grade myopia MYP3 locus reveals novel candidates UHRF1BP1L, PTPRR, and PPFIA2. Invest. Ophthalmol. Vis. Sci. 54, 2076–2086 (2013).
Gibson, J., Morton, N. E. & Collins, A. Extended tracts of homozygosity in outbred human populations. Hum. Mol. Genet. 15, 789–795 (2006).
Tanaka, M., Cabrera, V. M., González, A. M., Larruga, J. M., Takeyasu, T., Fuku, N. et al. Mitochondrial genome variation in eastern Asia and the peopling of Japan. Genome Res. 14, 1832–1850 (2004).
Schaaf, C. P., Wiszniewska, J. & Beaudet, A. L. Copy number and SNP arrays in clinical diagnostics. Annu. Rev. Genomics Hum. Genet. 12, 25–51 (2011).
Deciphering Developmental Disorders Study Large-scale discovery of novel genetic causes of developmental disorders. Nature 519, 223–228 (2015).
Acknowledgements
The cooperation of all the subjects that participated in this project, their relatives and all control individuals is greatly acknowledged. We thank all the members of the Japanese Array Consortium. We also thank Ayako Takahashi, Rumi Mori and Tomoko Ojima for technical assistance. This research was financially supported by the Joint Usage/Research Program of the Medical Research Institute, Tokyo Medical and Dental University; by grants-in-aid for Scientific Research on Priority Areas from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan; a grant (108808) from Core Research for Evolutional Science and Technology (CREST) of the Japan Science and Technology Corporation (JST); a grant (P06012) from New Energy and Industrial Technology Development Organization (NEDO); by grants-in-aid for Young Scientists (B) (23791155, 25830135) from Japan Society for the Promotion of Science (JSPS); and a Health Labour Sciences Research grant (201231015B) from the Ministry of Health, Labour and Welfare of Japan. Daniela Tiaki Uehara is supported by the MEXT scholarship program.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Uehara, D., Hayashi, S., Okamoto, N. et al. SNP array screening of cryptic genomic imbalances in 450 Japanese subjects with intellectual disability and multiple congenital anomalies previously negative for large rearrangements. J Hum Genet 61, 335–343 (2016). https://doi.org/10.1038/jhg.2015.154
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2015.154
This article is cited by
-
Copy number variation analysis in 189 Romanian patients with global developmental delay/intellectual disability
Italian Journal of Pediatrics (2022)
-
A missense variant in NUF2, a component of the kinetochore NDC80 complex, causes impaired chromosome segregation and aneuploidy associated with microcephaly and short stature
Human Genetics (2021)
-
Cytogenetic analysis of spontaneously discharged products of conception by array-based comparative genomic hybridization
SpringerPlus (2016)

{kind=link}
{kind=link}
{kind=link}