
Abstract
Two new chlorinated metabolites 4-chloro-1-hydroxy-3-methoxy-6-methyl-8-methoxycarbonyl-xanthen-9-one (1) and 2′-acetoxy-7-chlorocitreorosein (2), together with three known compounds (3–5), were obtained from the EtOAc extract of the endophytic fungus Penicillium citrinum HL-5126 isolated from the mangrove Bruguiera sexangula var. rhynchopetala collected in the South China Sea. Their structures were elucidated by the detailed analysis of comprehensive spectroscopic data. All compounds were evaluated for their antibacterial and topoisomerase I inhibitory activities. Compound 2 exhibited antibacterial activity against Vibrio parahaemolyticus with an MIC value of 10 μm.
Similar content being viewed by others
Introduction
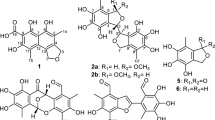
Marine organisms have attracted increasing attention from those searching for novel structural and biological diversity natural products in recent years. Especially, marine-derived natural halogenated compounds possess a variety of bioactivities such as cardiovascular, antibacterial, antifungal, cytotoxic, antiviral and insecticidal activities.1 For example, the brominated testafuran A was used as lead for treatment of cardiovascular disorders, merochlorins A and B were both potent inhibitors of methicillin-resistant Staphylococcus aureus (MRSA) and dankastatin C exhibited potent growth inhibition of P388 cell lines.2, 3, 4, 5 To date, more than 5000 halogenated natural products have been discovered,6, 7 these results have drawn the attention of both pharmaceutical and natural product chemists. In our search for new bioactive natural products from marine fungi in the South China Sea, we have found several bioactive compounds, including anthraquinone derivatives, cytochalasins, benzopyrans, stemphol sulfates and α-pyrone derivatives, from marine-derived fungus.8, 9, 10, 11, 12 Our previous investigation on the endophytic fungi Penicillium citrinum has resulted in the discovery of one new benzopyran derivative (2R*,4R*)-3,4-dihydro-5-methoxy-2-methyl-2H-1-benzopyran-4-ol and three new dihydroisocoumarin penicimarins G−I.10, 13 Further chemical investigation of the fermentation broth of P. citrinum resulted in the isolation of one new chlorinated xanthone 4-chloro-1-hydroxy-3-methoxy-6-methyl-8-methoxycarbonyl-xanthen-9-one (1) and one new chlorinated anthraquinone 2′-acetoxy-7-chlorocitreorosein (2), together with three known compounds, chloroisosulochrin dehydrate (3), citreorosein (4) and MT-1 (5) (Figure 1). All isolated metabolites (1–5) were evaluated for their antibacterial and topoisomerase I inhibitory activities. Herein we report the isolation, structure elucidation and biological activities of these compounds.

The structures of compounds 1–5.
Results and discussion
Isolation and identification of compounds
Compound 1 was isolated as a pale yellow powder, which has a molecular formula of C17H13ClO6, as determined by the HRESIMS, requiring 11 degrees of unsaturation and containing one chlorine atom. The 1H NMR spectrum of 1 (Table 1) was characterized by resonances consistent with one hydrogen-bonded phenol moiety at δH 12.66 (s, 1-OH), three aromatic methine protons at δH 7.45 (brs, H-5), 7.15 (brs, H-7) and 6.44 (s, H-2), two methoxy groups at δH 4.01 (s, 10-OMe) and 3.99 (s, 3-OMe), and a methyl group at δH 2.52 (s, 6-Me). The 13C NMR spectrum (Table 2) revealed 17 carbon signals, including two carbonyls δC (180.5 and 163.1), three aromatic methine carbons and nine aromatic quaternary carbons, together with two methoxy carbons at δC (57.9 and 54.2), and one methyl carbon at δC 23.0. Its 1H- and 13C-NMR spectroscopic data were closely resembled those of 3,14 except for the small shifting of the chemical shifts at 12 aromatic carbons in 1. In the HMBC spectrum, the correlations from the protons of 1-OH to C-1, C-1a and C-2, indicating that 1-OH was located at C-1, 3-OMe to C-3, 6-Me to C-5, C-6, C-7 and 10-OMe to C-10 indicating that the two methoxy groups and one methyl group were located at C-3, C-6 and C-10. Further HMBC correlations from H-2 to C-1a, C-3 and C-4, from H-5 to C-7, C-9a, C-10a and from H-7 to C-5, C-9a and C-10 indicated the position of the substituents in the structure. Chlorination of C-4 was found to be consistent with the C-4 chemical shift at δC 100.5. The 1H,1H-COSY, HMQC and HMBC spectra allowed the complete assignment for 1 (Figure 2). Thus, the structure of 1 was identified shown in Figure 1. We named compound 1 as 4-chloro-1-hydroxy-3-methoxy-6-methyl-8-methoxycarbonyl-xanthen-9-one.

Key HMBC correlations of compounds 1 and 2.
Compound 2 was isolated as a yellow powder, and it has a molecular formula of C18H13ClO7, as determined by the HRESIMS, requiring 12 degrees of unsaturation and containing one chlorine atom. The 1H NMR spectrum of 2 (Table 1) displayed one hydrogen-bonded phenol moiety at δH 12.93 (s, 1-OH), three aromatic methine protons at δH 7.59 (d, J =1.2 Hz, H-4), 7.31 (d, J=1.2 Hz, H-2) and 7.56 (s, H-5), one oxygen methylene group at δH 5.18 (s, H-1′), one methoxy group at δH 3.88 (s, 8-OMe) and a methyl group at δH 2.14 (s, H-3′). The 13C NMR spectrum (Table 1) revealed 18 carbon signals, including two carbonyls (δC 186.0 and 181.3, C-9 and C-10), three aromatic methine carbons, nine aromatic quaternary carbons. These spectroscopic features and the chemical shifts indicated that 2 has a highly substituted anthraquinone scaffold, and together with one methoxy carbon at δC 60.9, one oxygen methylene carbon at δC 64.2, and an acetyl group at δC 20.6 for methyl carbon and δC 170.1 for carbonyl carbon. In the HMBC spectrum, both H-4 and H-5 showed correlations to the carbonyl moiety at δC 181.3 (Figure 1), indicating the connectivity of these protons to C-4 and C-5, the correlations from 1-OH to C-1/1a/2 indicated the OH was linked to C-1, the correlation from 8-OMe to C-8 revealed that the methoxy group was located at C-8, the correlations from H-2 and H-4 to C-1′ indicated the oxygen methylene group was linked to C-3, and the correlations from H-1′ and 3′-Me to C-2′ indicated the acetyl group was linked at C-1′. Chlorination of C-7 was found to be consistent with the C-7 chemical shift at δC 122.6. The 1H,1H-COSY, HMQC and HMBC spectra allowed the complete assignment for 2 (Figure 2). Ultimately, the structure of new compound 2 was elucidated as 2′-acetoxy-7-chlorocitreorosein.
The structure of known 3–5 was identified by comparison of their 1H/13C NMR spectra with those in the literature.15, 16, 17 Compounds 1 and 3 were xanthones, and compounds 2, 4 and 5 were anthraquinones. The xanthones may be derived from the corresponding anthraquinones. The proposed biosynthesis pathway of the isolated compounds 1–5 is shown in Scheme 1.
Biological properties of compounds 1–5
The antibacterial activities of all the compounds were determined against four terrestrial pathogenic bacteria Escherichia coli (ATCC 25922), S. aureus (ATCC 25923), methicillin-resistant S. aureus (ATCC 33591), Bacillus cereus (ATCC 11778) and two marine pathogenic bacteria Vibrio parahaemolyticus (ATCC17802) and Vibrio alginolyticus (ATCC17749). Compounds 2 and 4 exhibited antibacterial activity against S. aureus with the same MIC values of 22.8 μm, respectively (Table 2). Compound 2 exhibited antibacterial activity against V. parahaemolyticus with the MIC value of 10 μm (Table 2). All compounds were also tested for their topoisomerase I inhibitory activities, however, these compounds showed no topoisomerase I inhibitory activity at the concentration of 500 μm.
Methods
Fungal materials
The fungal strain P. citrinum HL-5126 was isolated from the mangrove Bruguiera sexangula var. rhynchopetala collected in the South China Sea in August 2012. The strain was deposited in the Key Laboratory of Tropical Medicinal Plant Chemistry of Ministry of Education, College of Chemistry and Chemical Engineering, Hainan Normal University, Haikou, China, with the access code HL-5126. The fungal strain was cultivated in 30 l potato glucose liquid medium, 1 l Erlenmeyer flasks each containing 300 ml of medium for 100 Erlenmeyer flasks at 25 °C without shaking for 28 days. The potato glucose liquid medium contained 15 g of glucose and 30 g of sea salt in 1 l of potato infusion (potato infusion: 200 g slice potatoes thin were added to 1 l water, and boiled until soft for ~30 min. They were filtered through cheese cloth, and then the filtrate was hold to 1 l with distilled water).
Identification of fungus
The strain was identified as P. citrinum according to morphologic traits and molecular identification. Its 497 base pair ITS sequence had 99% sequence identity to that of P. citrinum (LP54362). The sequence data have been submitted to GenBank with the accession number KJ466981.
General experimental procedures
Octadecylsilyl silica gel (YMC, Kyoto, Japan; 12 nm–50 μm), silica gel (Qing Dao Hai Yang Chemical Group, Qing Dao, China; 200–300 mesh) and Sephadex LH-20 (GE) were used for column chromatography (CC). Precoated silica gel plates (Yan Tai Zi Fu Chemical Group, Yantai, China; G60, F-254) were used for TLC. 1H and 13C NMR spectra were recorded on a Bruker AV spectrometer (Bruker, Zurich, Switzerland) at 400 and 100 MHz. Chemical shifts δ are reported in p.p.m., using tetramethylsilane as internal standard, and coupling constants (J) are in Hz. HRESIMS spectra were obtained from a Micromass Q-TOF spectrometer and Thermo Scientific LTQ Orbitrap XL spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). UV spectra were recorded on a Beckman DU 640 spectrophotometer (Brea, CA, USA). IR spectra were recorded on a Nicolet 6700 spectrophotometer (Thermo Fisher Scientific).
Extraction and isolation
The fungal cultures were filtered through cheese cloth, and the filtrate was extracted with EtOAc (3 × 30 l, 24 h each). The organic extracts were concentrated in vacuo to yield an oily residue (22.0 g), which was subjected to silica gel CC (petroleum ether, EtOAc v/v, gradient 100:0–0:100) to generate six fractions (Fr. 1−Fr. 7), Fr. 3 (1.2 g) was isolated by CC on silica gel eluted with petroleum ether–EtOAc to obtain Fr. 3. 1 (390.0 mg), Fr. 3. 2 (410.0 mg) and Fr. 3.3 (200.0 mg). Fr. 3.3 was subjected to Sephadex LH-20 CC eluting with mixtures of Petroleum ether: CHCl3:MeOH (2:1:1), and further purified by using HPLC on an ODS semi-preparative column (Agilent C18, AgilentTechnologies, Santa Clara, CA, USA; 9.4 × 250 mm, 7 μm, 2 ml min−1) eluted with 85% MeOH/H2O to obtain compounds 1 (10.0 mg) and 3 (8.0 mg). Fr. 4 was isolated by CC on silica gel eluted with petroleum ether–EtOAc, and then subjected to Sephadex LH-20 CC eluting with mixtures of CHCl3–MeOH (1:1) to obtain Fr. 4. 1 (510.0 mg), Fr. 4. 2 (210.0 mg) and Fr. 4. 3 (340.0 mg). Fr. 4. 2 was subjected to repeated Sephadex LH-20 CC (CHCl3/MeOH, v/v, 1:1) and further purified by using HPLC on an ODS semi-preparative column (Agilent C18, 9.4 × 250 mm, 7 μm, 2 ml min−1) eluted with 80% MeOH/H2O to obtain compound 2 (9.0 mg). Fr. 4. 3 was subjected to repeated Sephadex LH-20 CC (CHCl3/MeOH, v/v, 1:1) and further purified by using HPLC on an ODS semi-preparative column (Agilent C18, 9.4 × 250 mm, 7 μm, 2 ml min−1) eluted with 75% MeOH/H2O to obtain compounds 4 (11.0 mg) and 5 (7.0 mg).
Physical properties of compounds 1 and 2
4-chloro-1-hydroxy-3-methoxy-6-methyl-8-methoxycarbonyl-xanthen-9-one (1): white amorphous powder. λmax (log ɛ) 238 (3.42), 262 (3.36), 296 (2.89), 385 (1.72) nm; IR (KBr) νmax 3440, 1740, 1610, 1280, 1240, 1210, 1112, 1082, 816 cm–1; 1H and 13C NMR see Table 1; HRESIMS m/z 349.0475 [M + H]+ (calcd for, C17H13ClO6 349.0479).
2′-acetoxy-7-chlorocitreorosein (2): yellow powder. λmax (log ɛ) 220 (4.02), 262 (3.80), 314 (4.12), 470 (3.37) nm; IR (KBr) νmax 3530, 2910, 1712, 1630, 1260, 1112, 1080 cm–1; 1H and 13C NMR see Table 1; HRESIMS m/z 775.0687 [2M + Na]+ (calcd for C36H26Cl2O14Na, 775.0597).
Biological assays
Antibacterial assays
Antibacterial activity was determined by the conventional broth dilution assay.18 Four terrestrial pathogenic bacteria E. coli, S. aureus, methicillin-resistant S. aureus, B. cereus and two marine pathogenic bacteria V. parahaemolyticus and V. alginolyticus were used, and ciprofloxacin was used as a positive control.
Topoisomerase I inhibitory assays
The enzyme topoisomerase I was from calf thymus. Relaxation activity of top I was performed as described by Bogurcu et al.,19 with some modification, using 10-hydroxycamptothecin as a positive control.

Proposed biosynthesis pathway of the isolated compounds 1–5.
Accession codes
References
Blunt, J. W., Copp, B. R., Keyzers, R. A., Munro, M. H. G & Prinsep, M. R. Marine natural products. Nat. Prod. Rep. 32, 116–211 (2015).
Akiyama, T. et al. Stimulators of adipogenesis from the marine sponge Xestospongia testudinaria. Tetrahedron 69, 6560–6564 (2013).
Sakoulas, G . et al. Novel bacterial metabolite merochlorin A demonstrates in vitro activity against multi–drug resistant methicillin–resistant Staphylococcus aureus. PLoS ONE 7, e29439 (2012).
Kaysser, L . et al. Merochlorins A–D, cyclic meroterpenoid antibiotics biosynthesized in divergent pathways with vanadium-dependent chloroperoxidases. J. Am. Chem. Soc. 134, 11988–11991 (2012).
Amagata, T. et al. Additional cytotoxic substances isolated from the sponge-–derived Gymnascella dankaliensis. Tetrahedron Lett. 54, 5960–5962 (2013).
Gribble, G. W. Naturally occurring organohalogen compounds−a comprehensive update (Springer-Verlag/Wien: New York, USA, 2010)..
Huang, H. B. et al. Halogenated anthraquinones from the marine–derived fungus Aspergillus sp. SCSIO F063. J. Nat. Prod. 75, 1346–1352 (2012).
Zhou, X. M. et al. Bioactive anthraquinone derivatives from the mangrove-derived fungus Stemphylium sp. 33231. J. Nat. Prod. 77, 2021–2028 (2014).
Zheng, C. J. et al. Bioactive phenylalanine derivatives and cytochalasins from the soft coral-derived fungus Aspergillus elegans. Mar. Drugs 11, 2054–2068 (2013).
Zheng, C. J. et al. A new benzopyrans derivatives from a mangrove-derived fungus Penicillium citrinum from the South China Sea. Nat. Prod. Res. 30, 821–825 (2016).
Zhou, X. M. et al. Two new stemphol sulfates from the mangrove endophytic fungus Stemphylium sp. 33231. J. Antibiot. 68, 501–503 (2015).
Zhou, X. M. et al. Antibacterial α-pyrone derivatives from a mangrove-derived fungus Stemphylium sp. 33231 from the South China Sea. J. Antibiot. 67, 401–403 (2014).
Huang, G. L. et al. Dihydroisocoumarins from the mangrove-derived fungus Penicillium citrinum. Mar. Drugs 14, E177 (2016).
Fujimoto, H., Inagaki, M., Satoh, Y., Yoshida, E. & Yamazaki, M. Monoamine oxidase-inhibitory components from an ascomycete, Coniochaeta tetraspora. Chem. Pharm. Bull. 44, 1090–1092 (1996).
Shimada, A., Takahashi, I., Kawano, T. & Kimura, Y. Chloroisosulochrin, chloroisosulochrin dehydrate, and pestheic acid, plant growth regulators, produced by Pestalotiopsis theae. Z. Naturforsch. B 56, 797–803 (2001).
Ma, W. L., Yan, C. Y., Zhu, J. H., Duan, G. Y. & Yu, R. M. Biotransformation of paeonol and emodin by transgenic crown galls of Panax quinquefolium. Appl. Biochem. Biotechnol. 160, 1301–1308 (2010).
Fujimoto, H., Fujimaki, T., Okuyama, E. & Yamazaki, M. Immunomodulatory constituents from an ascomycete, Microascus tardifaciens. Chem. Pharm. Bull. 47, 1426–1432 (1999).
Pierce, C. G. et al. A simple and reproducible 96-well plate-based method for the formation of fungal biofilms and its application to antifungal susceptibility testing. Nat. Protoc. 3, 1494–1500 (2008).
Bogurcu, N., Sevimli-Gur, C., Ozmen, B., Bedir, E. & Korkmaz, K. S. ALCAPs induce mitochondrial apoptosis and activate DNA damage response by generating ROS and inhibiting topoisomerase I enzyme activity in K562 leukemia cell line. Biochem. Biophys. Res. Commun. 409, 738–744 (2011).
Acknowledgements
This work was supported by the Foundation for the National Natural Science Foundation of China (No. 21462015 and 21662012), Marine Science and Technology Program of Hainan Province (No. 2015XH06), National Natural Science Foundation of Hainan Province (No. 20152034), Program for Innovative Research Team in University (IRT-16R19), Hainan Province Natural Science Foundation of Innovative Research Team Project (No. 2016CXTD007).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on The Journal of Antibiotics website
Supplementary information
Rights and permissions
About this article

Cite this article
He, KY., Zhang, C., Duan, YR. et al. New chlorinated xanthone and anthraquinone produced by a mangrove-derived fungus Penicillium citrinum HL-5126. J Antibiot 70, 823–827 (2017). https://doi.org/10.1038/ja.2017.52
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2017.52
This article is cited by
-
Fungal quinones: diversity, producers, and applications of quinones from Aspergillus, Penicillium, Talaromyces, Fusarium, and Arthrinium
Applied Microbiology and Biotechnology (2021)
-
A New Antibacterial Chlorinated Amino Acid Derivative from the Sponge-Derived Fungus Aspergillus sp. LS53
Chemistry of Natural Compounds (2020)
-
Endophytic Penicillium species and their agricultural, biotechnological, and pharmaceutical applications
3 Biotech (2020)
-
Two new secondary metabolites from a mangrove-derived fungus Cladosporium sp. JS1-2
The Journal of Antibiotics (2019)
