
Abstract
Obesity is associated with chronic inflammation of various tissues including visceral adipose tissue (VAT), which contributes to insulin resistance. T cells and macrophages infiltrate VAT in obesity and orchestrate this inflammation. Recently, we made the surprising discovery that B cells are important contributors to this process. Thus, some B cells and the antibodies they produce can promote VAT-associated and systemic inflammation, leading to insulin resistance. This report will focus on the properties of these B cells, and how they contribute to insulin resistance through T-cell modulation and production of pathogenic autoantibodies. Understanding the mechanisms by which B cells contribute to insulin resistance should lead to new antibody-based diagnostics and B-cell modulating therapeutics to manage this increasingly prevalent disease.
Similar content being viewed by others


Introduction
Insulin resistance is a state that precedes type 2 diabetes (T2D) in mice and humans and is a key feature of obesity-related metabolic syndrome, one of the world's fastest growing epidemics.1 Multiple factors contribute to insulin resistance, but inflammation of visceral adipose tissue (VAT) resulting in chronic release of pro-inflammatory cytokines is a major contributor.2, 3 Macrophage infiltration of VAT is a key event in the development of chronic inflammation leading to insulin resistance.4, 5, 6 Activation and polarization of macrophages in VAT are influenced by local immune cell cytokine production. In obesity, IFNγ-secreting CD8+ T cells and Th1 CD4+ T cells infiltrating VAT enhance the formation and pro-inflammatory functions of M1 macrophages.7, 8 Such macrophages in VAT produce IL-1β, IL-6 and TNFα, which can alter insulin receptor signaling in target tissues, directly contributing to insulin resistance. These cytokines are present at increased levels in the serum of obese individuals and contribute to systemic insulin resistance. By contrast, in lean individuals, VAT is dominated by IL-4- and IL-13-secreting Th2 CD4+ T cells, eosinophils, as well as IL-10- and TGF-β-producing CD4+ Foxp3+ T cells, which induce anti-inflammatory IL-10-secreting M2 or ‘alternatively activated’ macrophages.7, 9, 10, 11 Thus, the balance of macrophage polarization and ensuing VAT inflammation is directly influenced by T-cell functional status.
In addition to T cells, B cells also infiltrate VAT during diet-induced obesity, and only recently has their role in the development of insulin resistance been elucidated. B cells can modulate the immune system at multiple levels through antigen presentation, cytokine secretion and antibody production. Both systemic and local VAT-associated alterations in B-cell subsets, their cytokine profiles and the antibodies they produce are found in obesity and associated insulin resistance. These alterations in B cells contribute to T-cell cytokine release and macrophage polarization in VAT, and represent a new means by which the adaptive immune system exerts physiological control over systemic and VAT-associated inflammation and insulin resistance. This review will summarize the recent findings implicating B cells in the pathogenesis of insulin resistance.
B-cell subsets
B cells are important in the production of natural and specific antibodies, antigen presentation and modulation of T-cell activation. B cells consist of distinct subsets with differing surface phenotypes, functions and cytokine secretion profiles. They can be divided into two broad classes, B-1 or B-2 cells, and both types are found in VAT and can produce antibodies.
B-1 cells are enriched in mucosal tissues and body cavities, including the pleural, pericardial and peritoneal cavities. They can be classified further into B-1a cells, which are the main producers of natural IgM antibodies, and B-1b cells, which mediate adaptive humoral responses to T-independent antigens.12 B-1 cells circulate from the peritoneal cavity into VAT, mesenteric lymph nodes and spleen via a CXCL13-dependent process, especially upon breach of the intestinal barrier.13 In VAT, B-1 cells can localize to fat-associated lymphoid clusters, which consist of various mature and immature immune cells.14 B-1 cells express a less diverse but more polyreactive antibody repertoire compared with B-2 cells15 and are highly responsive to pathogen-associated molecules such as lipopolysaccharide.13 Thus, they provide crucial immunity to bacterial pathogens. For instance, B-1 cells express a unique antibody repertoire, including protective neutralizing IgM antibodies against phosphorylcholine on Streptococcus pneumoniae, that cross-react with membrane lipids on apoptotic cells and with modified forms of circulating lipids including oxidized low-density lipoprotein (oxLDL).16, 17 Such neutralizing IgM antibodies are considered to be protective against inflammation caused by oxLDL in atherosclerosis.18, 19 B-1 cells are also a major source of IL-10 and may exert anti-inflammatory effects via this cytokine.20
B-2 cells are conventional B cells found most frequently in secondary lymphoid organs.21 They can be subdivided into mature transitional cells (T1, T2 and T3) and mature follicular or marginal zone B cells.22 B-2 cells produce specific antibodies to T-dependent antigens. Depending on the cytokine environment, B-2 cells can differentiate into effector cells, which produce IL-2, as well as pro-inflammatory cytokines, such as IFNγ and IL-12, or anti-inflammatory and Th2-like cytokines, such as IL-10 and IL-4.23 B cells that specialize in producing large amounts of IL-10 have been termed ‘B10’ cells, some of which coexpress CD1d and CD5 and have the ability to exert significant influence on T-cell activation and some inflammatory responses.24
Pathophysiology of B cells in insulin resistance
B cells infiltrate VAT in diet-induced obesity
Both B-1 and B-2 cells are found in VAT. In C57BL/6 mice placed on a high-fat diet (HFD), B cells infiltrate adipose tissue within the first 3 weeks of diet initiation.25, 26 This B-cell infiltrate includes both B-1 and B-2 B cells. By 6–12 weeks on HFD, VAT-associated B cells demonstrate evidence of participation in an active immune response, including class switching to IgM+IgD− and IgG+ isotypes. In addition to localizing to fat-associated lymphoid clusters in obesity, B cells aggregate between and around stressed and dying adipocytes, likely sampling antigens from these cells and presenting their processed peptides to T cells. Indeed, B cells are often found in association with macrophages and T cells, forming the so-called ‘crown-like structures’ (CLS).2 CLS in the VAT of obese individuals are thought to be regions of cytokine and immune cell cross talk.
T-cell modulation
Our recent work has shown that some B cells can promote insulin resistance in obesity. B-cell-deficient (Bnull) mice on a HFD show improved glucose tolerance and insulin sensitivity despite weight gain, when compared with the control wild-type mice.26 One mechanism by which B-2 B cells promote insulin resistance is by interacting with T cells in an MHC-dependent manner to induce their secretion of pro-inflammatory IFNγ, a crucial cytokine in M1 macrophage polarization. Indeed, transfer of B cells lacking MHC into Bnull mice fails to worsen glucose tolerance, indicating the importance of cognate interactions with T cells in promoting insulin resistance. It is also possible that reciprocal interactions occur, with T cells influencing B cells in VAT, through induction of IgG class switching, possibly enhanced through CD40-CD40L interactions.27
Cytokine production
In addition to antigen presentation, B cells are also significant producers of cytokines that can modulate insulin resistance. IL-10 is typically considered an anti-inflammatory cytokine, and can be produced by B1 and B10 cells in mice and CD27+ IL-10-producing B cells in humans.28 B-cell-derived IL-10 has been shown to be a critical mediator in protection against inflammatory diseases such as experimental autoimmune encephalomyelitis.29, 30 In humans, decreased IL-10 production capacity is associated with metabolic syndrome and T2D.31 Consistently, B cells isolated from individuals with T2D secrete less IL-10 in response to TLR2, TLR4 and TLR9 agonists.32 As IL-10 can block Th1T-cell differentiation, it is possible that systemic and local B-cell-derived IL-10 in VAT holds Th1 cells in check, to maintain metabolic balance. Along with decreased production of IL-10, B cells have been shown to produce higher levels of IL-8, a pro-inflammatory chemokine, in individuals with T2D, but not IL-6 when compared with nondiabetic control subjects.32
Antibody production
In the spleens of diet-induced obese (DIO) mice, B cells show increased spontaneous production of IgG and decreased IgM production, consistent with the concept of obesity altering systemic immunity. In Akita mice, which develop hyperglycemia due to insulin misfolding, hyperglycemia can delay the production of IgM from LPS-stimulated splenocytes. Thus, hyperglycemia alone can alter B-cell antibody production.33 In response to HFD, sera and VAT from DIO mice show increased levels of pro-inflammatory IgG2c antibody, further evidence that class switching of antibody occurs as a direct consequence of obesity.26 Oral feeding of protein causes obese mice to develop IgG2a/c to protein antigens, whereas the same antigens elicit a less inflammatory IgG1 isotype in lean mice.34
Antibodies have direct pathogenic roles in autoimmune diseases such as systemic lupus and myasthenia gravis, but until recently there had been little or no evidence that autoantibodies contribute to the development of insulin resistance. Our recent findings challenge this notion, as transfer of serum IgG from DIO C57BL/6 mice into DIO B-cell-deficient recipients can promote insulin resistance.26 This effect requires exposure of both IgG donor and recipient mice to HFD, suggesting that possible diet-induced conditioning or induction of target antigens may be important.26 The effect of antibodies on insulin resistance occurs in an Fc-dependent manner, indicating that one potential mechanism of antibody-induced insulin resistance occurs via FcRγ activation of immune cells, most likely macrophages. Indeed, HFD IgG can prime VAT macrophages to produce TNFα both in vitro and in vivo.26 FcRs are also present on adipocytes, and it may be possible that antibodies bind directly to adipocytes to influence insulin resistance.35 Antibodies can also fix complement proteins, and thus additional information is needed to determine whether obesity-associated IgG has a role in the production of complement protein C3a, which binds its receptor C3aR on macrophages, both of which have recently been described as key mediators of insulin resistance.36
Association of an autoantibody signature with insulin resistance in humans
The identification of autoantibodies as mediators of insulin resistance predicts that certain targeted antigens will be associated with the insulin-resistant state. Indeed, in a cohort of 32 overweight to obese subjects, we found that insulin resistance or sensitivity is associated with distinct autoantibody clusters.26 Most of the antigens targeted in the insulin-resistant state are intracellular proteins, many of which are expressed in multiple tissues such as immune cells, pancreas, nerves, muscle or fat. Examples of these antigens include Golgi SNAP receptor complex member 1 (GOSR1), transcript variant 1, targeted in more than 70% of insulin-resistant male patients in one cohort.26 GOSR1 is part of the Golgi SNAP receptor complex and functions in protein trafficking between the endoplasmic reticulum and Golgi. More information is needed to determine how GOSR1 transcription is regulated and whether it varies in response to ER stress, a hallmark of insulin resistance.37
Another targeted antigen is glial fibrillary acidic protein (GFAP), to which 30–50% of patients with insulin resistance or T2D develop autoantibodies.26, 38 Whether the source of GFAP antigen responsible for inducing immunity originates in the central nervous system or other tissues, including the pancreas, remains an interesting question for future study.39 Other reports have shown that antibodies that inhibit endothelial cell function occur in more than 30% of type 2 diabetics, and correlate with vascular complications.40, 41, 42 Another study has shown that some autoantibodies responsible for insulin resistance can be elicited as a consequence of infection. For instance, following streptococcal infection, antibodies are produced to protein disulfide isomerase, and can contribute to insulin resistance.43 The role of natural IgM antibodies from B-1 cells, which have been shown to be protective in atherosclerosis, remains unknown in insulin resistance. As the connection between insulin resistance and adaptive immunity strengthens, the number of autoantibodies associated with insulin resistance will likely increase. Identification of targeted antigens in insulin resistance will be crucial for the development of new antibody-based diagnostics and possible future vaccination approaches to insulin resistance.
Potential therapies that target pathogenic B cells
Besides diet and exercise, the current medical management of obesity-related insulin resistance mainly includes drugs such as Metformin, which target altered insulin and glucose metabolism. Because of the prominent inflammatory component of metabolic syndrome, which fuels the development of insulin resistance, inflammatory cells and their products are attractive new targets for the treatment of this disorder. Drugs targeting IL-1β44 and anti-inflammatory drugs, such as salsalate,45 have shown promising results in clinical trials. Targeting pathogenic B cells represents another possible future means of therapy. In this regard, a B-cell-depleting anti-CD20 antibody (rituximab) is FDA approved for the treatment of rheumatoid arthritis, whereas anti-BLyS (B-lymphocyte stimulator or B-cell-activating factor), which targets a B-cell survival factor, has been approved for the treatment of systemic lupus. Conventional B-2 cells require BLyS for survival, maturation and activation,46 and thus anti-BlyS (belumimab) preferentially targets B-2 cells, which produce pathogenic IgG, leaving B-1 cells, which produce IL-10 and natural IgM, intact. In mice maintained on a HFD, early depletion of B cells with an anti-CD20 antibody has a marked therapeutic benefit in insulin resistance, with effects linked to reduced production of IFNγ and TNFα in VAT.26 Other B-cell-modulating agents include transmembrane activator, calcium modulator and cyclophilin ligand interactor fusion proteins or antibodies to or inhibitors of CD19, CD22, spleen tyrosine kinase and a proliferation-inducing ligand.
Additional therapeutic approaches relevant to B cells in insulin resistance may include FcR modulation or cell-based therapy using IL-10-producing anti-inflammatory B cells. The identification of antibody signatures linked to either insulin-resistant or -sensitive states also raises the fascinating question as to whether boosting immunity against distinct antigens may dampen disease and provide the basis for a future preventative vaccine against insulin resistance.
Summary
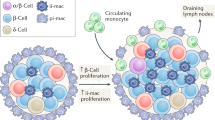
Our findings support a model in which B cells infiltrate VAT in obese individuals and associate with T cells and macrophages in CLS. B cells sample antigen from the surrounding tissues, possibly from dead adipocytes, and present peptides to VAT-associated CD4+ and CD8+ T cells, which produce IFNγ. In the presence of decreasing amounts of B-cell-derived IL-10, IFNγ polarizes nearby macrophages to an activated M1 phenotype. These M1 macrophages secrete large amounts of TNFα that acts in concert with IFNγ to cause insulin resistance. IL-8, produced by B cells, attracts additional inflammatory cells, including macrophages, to the site of VAT inflammation.47 B-cell-derived pathogenic IgG, including IgG2c, acts locally on macrophages in an Fc-dependent mechanism to further bolster TNFα production. IgG antibodies are also induced in spleens during obesity and may contribute systemically to insulin resistance in other target tissues. Figure 1 summarizes the roles of B cells in governing insulin resistance.

The role of B cells in VAT inflammation and insulin resistance. In the lean state, B cells produce IL-10, which promotes M2 macrophage polarization and insulin sensitivity. During obesity, B cells present insulin-resistance-associated antigens to T cells, leading to IFNγ production and M1 macrophage polarization. B cells undergo class switching to produce IgG2c with the potential to bind FcRs on macrophages and induce TNFα secretion. B cells also secrete pro-inflammatory chemokines such as IL-8, which may help recruit additional immune cells, including macrophages, to perpetuate the inflammation.
Conclusion
Our findings indicate that B cells have a critical role in the development of insulin resistance. These cells can promote insulin resistance through T-cell modulation and production of pathogenic antibodies. Some B cells secrete IL-10, and more work is needed to determine whether these B cells have a protective role in insulin resistance. The use of autoantibody-based assays represents a future avenue of diagnostic research in obesity, as does the modulation of B cells for potential therapy or vaccine development against this pervasive disease.
References
Franks PW, Hanson RL, Knowler WC, Sievers ML, Bennett PH, Looker HC . Childhood obesity, other cardiovascular risk factors, and premature death. N Engl J Med 2010; 362: 485–493.
Olefsky JM, Glass CK . Macrophages, inflammation, and insulin resistance. Annu Rev Physiol 2010; 72: 219–246.
Yuan M, Konstantopoulos N, Lee J, Hansen L, Li ZW, Karin M et al. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science 2001; 293: 1673–1677.
Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante Jr AW . Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 2003; 112: 1796–1808.
Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 2003; 112: 1821–1830.
Chawla A, Nguyen KD, Goh YP . Macrophage-mediated inflammation in metabolic disease. Nat Rev Immunol 2011; 11: 738–749.
Winer S, Chan Y, Paltser G, Truong D, Tsui H, Bahrami J et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat Med 2009; 15: 921–929.
Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med 2009; 15: 914–920.
Lumeng CN, Bodzin JL, Saltiel AR . Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest 2007; 117: 175–184.
Wu D, Molofsky AB, Liang HE, Ricardo-Gonzalez RR, Jouihan HA, Bando JK et al. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science 2011; 332: 243–247.
Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med 2009; 15: 930–939.
Haas KM, Poe JC, Steeber DA, Tedder TF . B-1a and B-1b cells exhibit distinct developmental requirements and have unique functional roles in innate and adaptive immunity to S pneumoniae. Immunity 2005; 23: 7–18.
Ha SA, Tsuji M, Suzuki K, Meek B, Yasuda N, Kaisho T et al. Regulation of B1 cell migration by signals through Toll-like receptors. J Exp Med 2006; 203: 2541–2550.
Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H et al. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature 2010; 463: 540–544.
Martin F, Kearney JF . B1 cells: similarities and differences with other B cell subsets. Curr Opin Immunol 2001; 13: 195–201.
Kearney JF . Immune recognition of OxLDL in atherosclerosis. J Clin Invest 2000; 105: 1683–1685.
Binder CJ, Silverman GJ . Natural antibodies and the autoimmunity of atherosclerosis. Springer Semin Immunopathol 2005; 26: 385–404.
Ait-Oufella H, Herbin O, Bouaziz JD, Binder CJ, Uyttenhove C, Laurans L et al. B cell depletion reduces the development of atherosclerosis in mice. J Exp Med 2010; 207: 1579–1587.
Kyaw T, Tay C, Khan A, Dumouchel V, Cao A, To K et al. Conventional B2 B cell depletion ameliorates whereas its adoptive transfer aggravates atherosclerosis. J Immunol 2010; 185: 4410–4419.
Mauri C, Ehrenstein MR . The ‘short’ history of regulatory B cells. Trends Immunol 2008; 29: 34–40.
Allman D, Pillai S . Peripheral B cell subsets. Curr Opin Immunol 2008; 20: 149–157.
Lund FE . Cytokine-producing B lymphocytes-key regulators of immunity. Curr Opin Immunol 2008; 20: 332–338.
Harris DP, Haynes L, Sayles PC, Duso DK, Eaton SM, Lepak NM et al. Reciprocal regulation of polarized cytokine production by effector B and T cells. Nat Immunol 2000; 1: 475–482.
Yanaba K, Bouaziz JD, Haas KM, Poe JC, Fujimoto M, Tedder TF . A regulatory B cell subset with a unique CD1dhiCD5+ phenotype controls T cell-dependent inflammatory responses. Immunity 2008; 28: 639–650.
Duffaut C, Galitzky J, Lafontan M, Bouloumie A . Unexpected trafficking of immune cells within the adipose tissue during the onset of obesity. Biochem Biophys Res Commun 2009; 384: 482–485.
Winer DA, Winer S, Shen L, Wadia PP, Yantha J, Paltser G et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med 2011; 17: 610–617.
Poggi M, Engel D, Christ A, Beckers L, Wijnands E, Boon L et al. CD40L deficiency ameliorates adipose tissue inflammation and metabolic manifestations of obesity in mice. Arterioscler Thromb Vasc Biol 2011; 31: 2251–2260.
Duddy M, Niino M, Adatia F, Hebert S, Freedman M, Atkins H et al. Distinct effector cytokine profiles of memory and naive human B cell subsets and implication in multiple sclerosis. J Immunol 2007; 178: 6092–6099.
Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM . B cells regulate autoimmunity by provision of IL-10. Nat Immunol 2002; 3: 944–950.
Matsushita T, Yanaba K, Bouaziz JD, Fujimoto M, Tedder TF . Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J Clin Invest 2008; 118: 3420–3430.
van Exel E, Gussekloo J, de Craen AJ, Frolich M, Bootsma-Van Der Wiel A, Westendorp RG . Low production capacity of interleukin-10 associates with the metabolic syndrome and type 2 diabetes: the Leiden 85-Plus Study. Diabetes 2002; 51: 1088–1092.
Jagannathan M, McDonnell M, Liang Y, Hasturk H, Hetzel J, Rubin D et al. Toll-like receptors regulate B cell cytokine production in patients with diabetes. Diabetologia 2010; 53: 1461–1471.
Nikolajczyk BS, Jagannathan-Bogdan M, Shin H, Gyurko R . State of the union between metabolism and the immune system in type 2 diabetes. Genes Immun 2011; 12: 239–250.
Mito N, Kaburagi T, Yoshino H, Imai A, Sato K . Oral-tolerance induction in diet-induced obese mice. Life Sci 2006; 79: 1056–1061.
Palming J, Gabrielsson BG, Jennische E, Smith U, Carlsson B, Carlsson LM et al. Plasma cells and Fc receptors in human adipose tissue—lipogenic and anti-inflammatory effects of immunoglobulins on adipocytes. Biochem Biophys Res Commun 2006; 343: 43–48.
Mamane Y, Chung Chan C, Lavallee G, Morin N, Xu LJ, Huang J et al. The C3a anaphylatoxin receptor is a key mediator of insulin resistance and functions by modulating adipose tissue macrophage infiltration and activation. Diabetes 2009; 58: 2006–2017.
Hotamisligil GS . Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010; 140: 900–917.
Gomez-Tourino I, Camina-Darriba F, Otero-Romero I, Rodriguez MA, Hernandez-Fernandez A, Gonzalez-Fernandez A et al. Autoantibodies to glial fibrillary acid protein and S100beta in diabetic patients. Diabet Med 2010; 27: 246–248.
Winer S, Tsui H, Lau A, Song A, Li X, Cheung RK et al. Autoimmune islet destruction in spontaneous type 1 diabetes is not beta-cell exclusive. Nat Med 2003; 9: 198–205.
Fredrikson GN, Anand DV, Hopkins D, Corder R, Alm R, Bengtsson E et al. Associations between autoantibodies against apolipoprotein B-100 peptides and vascular complications in patients with type 2 diabetes. Diabetologia 2009; 52: 1426–1433.
Zimering MB, Pan Z . Autoantibodies in type 2 diabetes induce stress fiber formation and apoptosis in endothelial cells. J Clin Endocrinol Metab 2009; 94: 2171–2177.
Hempel P, Karczewski P, Kohnert KD, Raabe J, Lemke B, Kunze R et al. Sera from patients with type 2 diabetes contain agonistic autoantibodies against G protein-coupled receptors. Scand J Immunol 2009; 70: 159–160.
Aran A, Weiner K, Lin L, Finn LA, Greco MA, Peppard P et al. Post-streptococcal auto-antibodies inhibit protein disulfide isomerase and are associated with insulin resistance. PLoS One 2010; 5: e12875.
Larsen CM, Faulenbach M, Vaag A, Volund A, Ehses JA, Seifert B et al. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med 2007; 356: 1517–1526.
Goldfine AB, Fonseca V, Jablonski KA, Pyle L, Staten MA, Shoelson SE . The effects of salsalate on glycemic control in patients with type 2 diabetes: a randomized trial. Ann Intern Med 2010; 152: 346–357.
Rauch M, Tussiwand R, Bosco N, Rolink AG . Crucial role for BAFF-BAFF-R signaling in the survival and maintenance of mature B cells. PLoS One 2009; 4: e5456.
Waugh DJ, Wilson C . The interleukin-8 pathway in cancer. Clin Cancer Res 2008; 14: 6735–6741.
Acknowledgements
This work was funded by grants from the Canadian Institutes of Health Research (DW, Grant 119414) and the National Institutes of Health (EE, Grants CA141468 and DK082537). Publication of this supplement was partially supported by Nutrilite Health Institute with an unrestricted educational contribution to Stanford Prevention Research Center.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Winer, D., Winer, S., Shen, L. et al. B lymphocytes as emerging mediators of insulin resistance. Int J Obes Supp 2 (Suppl 1), S4–S7 (2012). https://doi.org/10.1038/ijosup.2012.2
Published:
Issue Date:
DOI: https://doi.org/10.1038/ijosup.2012.2
Keywords
This article is cited by
-
Microenvironment of Immune Cells Within the Visceral Adipose Tissue Sensu Lato vs. Epicardial Adipose Tissue: What Do We Know?
Inflammation (2018)
-
BET bromodomain proteins and epigenetic regulation of inflammation: implications for type 2 diabetes and breast cancer
Cellular and Molecular Life Sciences (2017)
