
Abstract
We describe a novel autosomal recessive form of mitochondrial disease in a child with short stature, poor weight gain, and mild dysmorphic features with highly suspected Fanconi anemia due to a mutation in COX4I1 gene. Whole Exome Sequencing was performed then followed by Sanger confirmation, identified a K101N mutation in COX4I1, segregating with the disease. This nuclear gene encodes the common isoform of cytochrome c oxidase (COX) subunit 4 (COX 4-1), an integral regulatory part of COX (respiratory chain complex IV) the terminal electron acceptor of the mitochondrial respiratory chain. The patient’s fibroblasts disclosed decreased COX activity, impaired ATP production, elevated ROS production, decreased expression of COX4I1 mRNA and undetectable (COX4) protein. COX activity and ATP production were restored by lentiviral transfection with the wild-type gene. Our results demonstrate the first human mutation in the COX4I1 gene linked to diseases and confirm its role in the pathogenesis. Thus COX4I1 mutations should be considered in any patient with features suggestive of this diagnosis.
Similar content being viewed by others

Introduction
Mitochondrial cytochrome c oxidase (COX, complex IV) is the terminal electron acceptor of the mitochondrial respiratory chain catalyzing the transfer of electrons from ferrocytochrome c to molecular oxygen contributing to the electrochemical gradient utilized by ATPsynthase to form ATP. The mammalian COX is a dimeric multi-subunit complex composed of 13 subunits. The three mitochondrial-encoded subunits comprise the catalytic core, while the reminding subunits, encoded by the nuclear DNA perform regulatory functions.1 COX deficiency (OMIM # 220110) is prevalent among mitochondrial diseases, presenting with a marked clinical heterogeneity ranging from fatal neonatal, lactic acidosis to adult myopathy affecting numerous organs including brain, heart, muscle, liver, hematopoesis and pancreas. Mutations in more than 30 genes, in both mitochondrial and nuclear DNA, affecting either structural subunits of the enzyme or proteins involved in its biogenesis were associated with human disease.2 To date no disorder linked to COX4I1, encoding the common isoform of cytochrome c oxidase subunit 4 isoform 1 (COX 4-1) has been reported. Here we describe a novel COX defect in child with short stature, poor weight gain, and mild dysmorphic features, with features of Fanconi anemia due to a homozygous mutation in COX4I1.
Subjects and methods
Subject
The patient is a female child who was referred from Gaza Strip at the age of 3 2/12 years for evaluation of failure to thrive. She was a product of full-term normal vaginal delivery with birth weight of 2.8 kg after an uneventful pregnancy. Parents are first cousins with another two healthy children and negative family history of the same condition. She was noted by her parents since the age of 4 months to have poor weight gain. Routine tests at that time did not reveal any explanation of her poor weight gain. Her condition remained the same and at the age of 2 years she was diagnosed as failure to thrive of undetermined cause after inpatient evaluation at a hospital in Gaza Strip. Her psychosocial and motor developmental mile stones were within normal limits. No history of seizures. Brain MRI was performed and found normal without any indication of mitochondrial disease
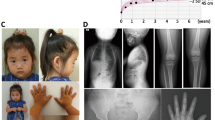
Her growth parameters at the age of 3 2/12 showed: weight of 8.31 kg (less than −3 SD); height of 80 cm (less than −3 SD) and occipitofrontal circumference of 46 cm (just at −2 SD). Her physical examination revealed a thin looking child with relative large head and normal systemic examination, except for minor dysmorphic features including prominent nasal bridge, frontal bossing, and short 5th fingers bilaterally (Figure 1). No pigmentation abnormalities were detected and her neurological examination was normal. In blood and plasma, ammonia, lactic acid, amino acid, B12 levels were all normal.

Patient features. The figure shows photographs of the patients’ facial (a) and hand (b) features at the age of 5 years (with parental permission).
Urinary organic acids analysis was unremarkable. CBC count was within normal range and there was no pancytopenia. Investigations were positive only for RBC macrocytosis and delayed bone age. The diagnosis of Fanconi anemia was suspected and peripheral blood karyotyping with Diepoxybutane (DEB)-induced chromosomal breakage study was requested. The result showed normal female karyotype and 29% of the cells showed ≥1 break/cell. This supported the clinical suspicion of Fanconi anemia and whole exome sequencing was requested to confirm the molecular genetic cause of her disease.
Methods
Whole exome analysis
Exonic sequences were enriched in the DNA sample of the patient using SureSelect Human All Exon 50 Mb Kit V4 (Agilent Technologies, Santa Clara, CA, USA). Sequences were determined by HiSeq2500 (Illumina, San Diego, CA, USA) as 100-bp paired-end runs. Data analysis including read alignment and variant calling was performed by DNAnexus software (Palo Alto, CA, USA) using the default parameters with the human genome assembly hg19 (GRCh37) as reference. Parental consent was given for DNA studies.
Tissue cultures
Primary skin fibroblasts (obtained with informed consent) were maintained in a permissive high glucose-DMEM medium containing 4.5 g/l glucose supplemented with 15% fetal calf serum, L-glutamine, pyruvate and 50 μg/ml uridine, at 37 °C in 5% CO2. For radical oxygen species (ROS) production cells were also cultivated in glucose-free DMEM medium (Biological Industries, Kibbutz Beit Haemek, Israel) supplemented with 10% dialyzed fetal calf serum and 5 mM galactose (GAL).
Functional studies
Mitochondrial ATP and ROS were measured as we have recently described.3 ATP production was measured in digitonin-permeabilized cells in the presence of glutamate and malate in microtiter wells essentially as we have recently described ATP was measured by luciferin–luciferase using ATPliteTM luminescence assay system according to the manufacturer’s instructions (Perkin Elmer, Waltham, MA, USA). ROS production was measured in regular and restrictive galactose-free medium by 2',7'-dichlorodihydrofluorescein diacetate. Results were normalized for cell content measured with methylene blue.
Enzymatic activities of cytochrome c oxidase (COX, complex IV) and citrate synthase (CS), a mitochondrial marker enzyme, were determined by spectrophotometry in fibroblast homogenates as we have previously described.4
Western Blot
Western blot analysis was performed after separation of isolated fibroblasts mitochondria by urea-sodiumdodecylsulfate-polyacrylamide gel electrophoresis (Urea-SDS-PAGE) as described.5The membrane was sequentially exposed to the following mouse monoclonal antibodies (MitoSciences Molecular Probes, Eugene, OR, USA): COX subunit 1 (catalog #A-6403), COX subunit 2 (catalog# A-6404), COX subunit 4 (catalog #A-6431), Complex I subunit NDUFA9 (catalog #MS111) Complex II 70 KDa subunit (catalog #A-11142) overnight followed by a 2-h incubation with peroxidase-conjugated goat-anti mouse antibodies (Jackson Immuno Research laboratories, West Grove, PA, USA) (catalog # 115-035-062)and visualized by enhanced chemiluminescence detection using the EZ-ECL kit (Biological Industries, Kibbutz Beit Ha’emek, Israel). Band intensities were estimated by ImageJ http://imagej.nih.gov/ij (National Institute of Health, Bethesda, MD, USA).
Real-time quantitative RT-PCR
Total RNA was isolated from with Tri-Reagent (Telron, Israel) and c-DNA was generated (ImPromII, Promega, Madison, WI, USA). Real-time PCR (RT-PCR) was performed using Fast SYBR GreenMaster Mix and the ABI PRISM7900HT sequence detection system (Applied Biosystems, Foster City, CA, USA) using primers for COXIV1 and for normalization GAPDH (primer sequences are provided in Supplementary Table 2)
Wild-type (wt) cDNA complementation
A lenti-viral vector containing the wild-type human hCOX4I1 gene from Cyagen (Cyagen Biosciences, Santa Clara, CA, USA) pLV[Exp]-Puro-CMV>hCOX4I1 was co-transfected with pLP1, pLP2 and pLP/VSVG plasmids with Lipofectamine-2000, according to the manufacturer’s instructions (Vira-Power, Lentiviral expression system, Invitrogen, Life Technologies, Grand Island, NY, USA) into 293FT cells. Patient’s fibroblasts were subsequently infected with the hCOX4I1 lentiviral supernatant overnight or with a control vector, in the presence of polybrene. Stably transduced cells obtained by 2 weeks of puromycin selection.
Results
Exome analysis of the patient yielded 72.7 million mapped reads with a mean coverage of X130. Following alignment and variant calling, we performed a series of filtering steps. These included removing variants which were called less than X8, were off-target, heterozygous, synonymous, had MAF>1% at ExAC (Exome Aggregation Consortium, Cambridge, MA, USA (URL: http://exac.broadinstitute.org)) or MAF>2% at the Hadassah in-house database (~900 ethnic matched exome analyses). Sixteen variants survived the filtering (Supplementary Table S1) and after consideration of conservation, frequency, and functional prediction, we focused on chr16: g.85839400_85839401delinsTT, NM_001861.3:c.303_304delinsTT, p.(Lys101_Thr102delinsAsnSer) in the COX4I1 gene. Since only Lys-101 was conserved throughout evolution (Figure 2), we refer to the variant as K101N. The variant data and phenotypes are submitted, and will be available at https://databases.lovd.nl/shared/individuals/COX4I1 (patient ID # 102112).

Evolutionary conservation of Lys-101 in the COX4I1 gene. The mutated conserved residue is highlighted.
The variant, located in exon 4 (numbered as in NG_042280.1), was deemed to affect function in silico by PolyPhen-2 http://genetics.bwh.harvard.edu/pph2/) and by Mutation Taster (http://www.mutationtaster.org/) predicted to be disease causing with a score above 0.999 with possible splice site changes.
The variant segregated in the family and was absent from the >60 000 exome analyses of healthy individuals deposited at ExAC website.
The patient’s fibroblasts reflected the phenotype as COX activity was significantly decreased (9.1 nmol/min/mg compared to controls 36.9±10 nmol/min/mg); this was also evident when normalized to citrate synthase showing a 66% decrease (Figure 3a). In accord, a 40% decrease in mitochondrial ATP production was observed (Figure 3b). Real-time quantitative RT-PCR in fibroblasts disclosed a marked 85% decrease in COX4I1 expression, compared to control cells when normalized to GAPDH. ROS production was elevated by 30% on galactose-free medium while no significant elevation was detected in glucose medium. Western blot analysis revealed undetectable COX 4 protein while complexes I and II were comparable to controls (Figure 4a). Notably, when normalized to complex II, COX subunits 1 and 2 were decreased to 50 and 25% of controls respectively, while complex I remained largely intact (85% of control) (Figure 4b).

COX enzymatic activity and ATP production in patient’s fibroblasts. Fibroblast from controls n=3, patient (Pt), patient cells transformed with a control lenti-virus (Pt+CV) and patient cells transformed with a lenti virus containing the wild-type COX 4I1 (Pt+ COX 4I1) were assayed for COX enzymatic activity (a), expressed as COX normalized to citrate synthase activity (U COX/U CS) and for ATP production (b), expressed as relative luminescence (RLU) normalized to cell content (A620). *P<0.05.

Western blot analysis. Twenty micrograms protein per slot obtained from mitochondria, isolated from patient (Pt) and controls (n=3) fibroblasts were subjected to Urea-SDS-PAGE and Western blot with antibodies against complexes I (CI), II (CII) and subunits 1, 2 and 4 of complex IV (COX) and visualized by ECL (A). The graph depicts intensities normalized to CII.
Complementation with wt COX4I1 gene by stable lentiviral transduction significantly improved both COX (Figure 3a) activity and ATP production, when compared to control vector (Figure 3b). Taken together, the above results confirm that the K101N variant is disease-causing in this specific context.
Discussion
Different from other respiratory chain complexes, several COX subunits occur in different isoforms and the two isoforms of COX subunit 4 were identified in 2001 by Hüttemann et al.6 Isoform 1 is the largest nuclear-encoded subunit and is ubiquitously expressed. It contains transmembrane helix domain in the middle with two extra membrane domains at both ends and has an allosteric regulatory, inhibitory function on COX activity mediated by ATP/ADP ratios and cAMP-dependent phosphorylation. Bacteria generally lack the COX subunit 4 and therefore allosteric ATP inhibition is absent.2 The K101N variant is located in a conserved residue in the transmembrane helix domain, which participates in the interaction with the catalytic COX subunits I and II which could explain their decreased abundance in the patients fibroblasts and obviously the decreased COX activity (Figure 3).7 The decreased mRNA expression could possibly be attributed to aberrant splicing and the exact nature of this will be subject to future investigations.
To the best of our knowledge, no human disease has been reported yet due to disease-causing variants in this gene. Notably Shteyer et al8 reported four Palestinian patients with exocrine pancreatic insufficiency, dyserythropoeitic anemia, and calvarial hyperostosis due to a disease-causing variant in the COX4I2 gene encoding the isoform COX4-2 which is expressed mainly in lung, induced under hypoxia.2, 8 Apart from these, only a few other nuclear encoded structural COX subunits have been reported to be linked to human disease, namely COX6A1 which is associated with the mixed form of Charcot–Marie–Tooth disease;9 COX6B1 causing severe encephalomyopathy,10, 11 COX7B linked to a skin lesions and microphthalmia12 and recently COX8A was reported to cause Leigh syndrome.13 Thus the spectrum of phenotypes of these disorders is extremely variable also including anemia. However, none of these mimicked Fanconi anemia.
Fanconi anemia is a rare inherited syndrome with diverse clinical symptoms including developmental defects, short stature, bone marrow failure, and a high risk of malignancies14 At presentation, patients with Fanconi anemia may have: (1) typical physical anomalies and abnormal hematologic findings (majority of the patients); (2) normal physical features but abnormal hematologic findings (about one-third of patients); or (3) physical anomalies and normal hematologic findings (unknown percentage).14
Fanconi anemia is caused by mutations in many genes and within the past 2 years, and after the discovery of five new loci, the total number of disease-causing genes has increased to 21 (FANCA-FANCV).15 In our patient, the suspected diagnosis of Fanconi anemia was based mainly on short stature, poor weight gain, and minor dysmorphic facial features. The clinical suspicion was highly supported by the abnormal DEB-induced chromosomal breakage study.16 Notably, oxidative stress and mitochondrial dysfunction has previously been implied in Fanconi anemia.17, 18, 19, 20 Specifically, expression studies in Fanconi anemia bone marrow cells disclosed changes in genes involved in the mitochondrial respiratory chain17 and FANCA fibroblasts and blood cells demonstrated defective respiration via Complex I.19 It was recently also demonstrated that defective mitochondrial energetic function compels Fanconi anemia cells to glycolytic metabolism.21 This is in accord with our results showing elevated ROS production from fibroblasts grown in restrictive, glucose-free medium where the respiratory chain is the sole cellular energy provider.
In the present case, whole exome sequencing was requested in order to determine the definite molecular genetic defect with focus on Fanconi anemia genes. The result did not show any exonic variants in any of the known Fanconi anemia genes; however, there was a predicted homozygous disease-causing variant in the COX4I1 gene. As routine metabolic workup was normal (which does neither confirm nor exclude COX deficiency22), functional studies were required and showed evidence of defective function of this gene confirming its role in the pathogenesis of the disease. Nevertheless, presently our findings do not fully explain the pathomechanistic link between a genetic, mitochondrial COX defect, and the Fanconi anemia-like presentation. Retrospective investigation of erythropoiesis in molecularly defined COX deficiencies additionally to COX4I1and COX4I28 and in animal models23 could contribute to the understanding of this entity.
In summary, our findings describe the first disease-causing variant in the COX4I1 gene and confirm its role in the pathogenesis of disease in the reported case. As the affected child is highly suspected to have Fanconi anemia, sequencing of this gene should be considered in any child with features suggestive of this diagnosis. COX deficiency due to a pathogenic variant in COX4I1 expands the genetic sub-types in this disease.
References
Kadenbach B, Hüttemann M : The subunit composition and function of mammalian cytochrome c oxidase. Mitochondrion 2015; 24: 64–76.
Rak M, Bénit P, Chrétien D, Bouchereau J, Schiff M, El-Khoury R, Tzagoloff A, Rustin P : Mitochondrial cytochrome c oxidase deficiency. Clin Sci (Lond) 2016; 130: 393–407.
Yu-Wai-Man P, Soiferman D, Moore DG, Burté F, Saada A : Evaluating the therapeutic potential of idebenone and related quinone analogues in Leber hereditary optic neuropathy. Mitochondrion 2017, e-pub ahead of print 16 January 2017; doi:10.1016/j.mito.2017.01.004.
Saada A, Bar-Meir M, Belaiche C, Miller C, Elpeleg O : Evaluation of enzymatic assays and compounds affecting ATP production in mitochondrial respiratory chain complex I deficiency. Anal Biochem 2004; 335: 66–72.
Darley-Usmar VM, Capaldi RA, Takamiya S et al: Reconstitution and molecular analysis of the respiratory chain; in Darley-Usmar VM, Rickwood D, Wilson MT (eds): Mitochondria a practical approach. Washington, DC: IRL press, 1987, pp 113–152.
Hüttemann M, Kadenbach B, Grossman LI : Mammalian subunit IV isoforms of cytochrome c oxidase. Gene 2001; 267: 111–123.
Tsukihara T, Aoyama H, Yamashita E et al: The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8A. Science 1996; 272: 1136–1144.
Shteyer E, Saada A, Shaag A et al: Exocrine pancreatic insufficiency, dyserythropoeitic anemia, and calvarial hyperostosis are caused by a mutation in the COX4I2 gene. Am J Hum Genet 2009; 84: 412–417.
Tamiya G, Makino S, Hayashi M, Abe A et al: A mutation of COX6A1 causes a recessive axonal or mixed form of Charcot-Marie-Tooth disease. Am J Hum Genet 2014; 95: 294–300.
Massa V, Fernandez-Vizarra E, Alshahwan S et al: Severe infantile encephalomyopathy caused by a mutation in COX6B1, a nucleus-encoded subunit of cytochrome c oxidase. Am J Hum Genet 2008; 82: 1281–1289.
Abdulhag UN, Soiferman D, Schueler-Furman O et al: Mitochondrial complex IV deficiency, caused by mutated COX6B1, is associated with encephalomyopathy, hydrocephalus and cardiomyopathy. Eur J Hum Genet 2015; 23: 159–164.
Indrieri A, van Rahden VA, Tiranti V et al: Mutations in COX7B cause microphthalmia with linear skin lesions, an unconventional mitochondrial disease. Am J Hum Genet 2012; 91: 942–949.
Hallmann K, Kudin AP, Zsurka G et al: Loss of the smallest subunit of cytochrome c oxidase, COX8A, causes Leigh-like syndrome and epilepsy. Brain 2016; 139: 338–345.
Freedman MH, Dror Y : The inherited pancytopenias; in: Kliegman R, Stanton B, St Geme J, Schor N, Behrman R (eds). Nelson Textbook of Pediatrics. Elsevier: Amsterdam, NL, 2016, 20th edn, pp 2362–2370.
Mamrak NE, Shimamura A, Howlett NG : Recent discoveries in the molecular pathogenesis of the inherited bone marrow failure syndrome Fanconi anemia. Blood Rev 31: 93–99.
Oostra AB, Nieuwint AW, Joenje H, de Winter JP : Diagnosis of fanconi anemia: chromosomal breakage analysis. Anemia 2012; 2012: 238731.
Pagano G, Talamanca AA, Castello G et al: Bone marrow cell transcripts from Fanconi anemia patients reveal in vivo alterations in mitochondrial, redox and DNA repair pathways. Eur J Haematol 2013; 91: 141–151.
Kumari U, Ya Jun W, Huat Bay B, Lyakhovich A : Evidence of mitochondrial dysfunction and impaired ROS detoxifying machinery in Fanconi anemia cells. Oncogene 2014; 33: 165–172.
Ravera S, Vaccaro D, Cuccarolo P et al: Mitochondrial respiratory chain Complex I defects in Fanconi anemia complementation group A. Biochimie 2013; 95: 1828–1837.
Pagano G, Talamanca AA, Castello G et al: From clinical description, to in vitro and animal studies, and backward to patients: oxidative stress and mitochondrial dysfunction in Fanconi anemia. Free Radic Biol Med 2013; 58: 118–125.
Cappelli E, Cuccarolo P, Stroppiana G et al: Defects in mitochondrial energetic function compels Fanconi Anaemia cells to glycolytic metabolism. Biochim Biophys Acta 2017; 1863: 1214–1221.
Alban C, Fatale E, Joulani A, Ilin P, Saada A : The Relationship between mitochondrial respiratory chain activities in muscle and metabolites in plasma and urine: a retrospective study. J Clin Med 2017; 31.
Diaz F : Cytochrome c oxidase deficiency: patients and animal models. Biochim Biophys Acta 2010; 1802: 100–110.
Acknowledgements
AS is supported by the Pakula family via AFHU. Elena Fatale is acknowledged for technical support.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Abu-Libdeh, B., Douiev, L., Amro, S. et al. Mutation in the COX4I1 gene is associated with short stature, poor weight gain and increased chromosomal breaks, simulating Fanconi anemia. Eur J Hum Genet 25, 1142–1146 (2017). https://doi.org/10.1038/ejhg.2017.112
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2017.112
This article is cited by
-
Personalized Protein-Protein Interaction Networks Towards Unraveling the Molecular Mechanisms of Alzheimer’s Disease
Molecular Neurobiology (2024)
-
Mitochondrial complex III deficiency drives c-MYC overexpression and illicit cell cycle entry leading to senescence and segmental progeria
Nature Communications (2023)
-
Genetic analysis of a Fanconi anemia case revealed the presence of FANCF mutation (exon 1;469>C-T) with implications to develop acute myeloid leukemia
Molecular Biology Reports (2023)
-
Mapping drug-target interactions and synergy in multi-molecular therapeutics for pressure-overload cardiac hypertrophy
npj Systems Biology and Applications (2021)
-
Cytochrome c oxidase deficiency, oxidative stress, possible antioxidant therapy and link to nuclear DNA damage
European Journal of Human Genetics (2018)
