
Abstract
Biallelic PMS2 mutations are responsible for more than half of all cases of constitutional mismatch repair deficiency (CMMRD), a recessively inherited childhood cancer predisposition syndrome. The mismatch repair gene PMS2 is partly embedded within one copy of an inverted 100-kb low-copy repeat (LCR) on 7p22.1. In an individual with CMMRD syndrome, PMS2 was found to be homozygously inactivated by a complex chromosomal rearrangement, which separates the 5′-part from the 3′-part of the gene. The rearrangement involves sequences of the inverted 100-kb LCR and a human endogenous retrovirus element and may be associated with an inversion that is indistinguishable from the known inversion polymorphism affecting the ~0.7-Mb sequence intervening the LCR. Its formation is best explained by a replication-based mechanism (RBM) such as fork stalling and template switching/microhomology-mediated break-induced replication (FoSTeS/MMBIR). This finding supports the hypothesis that the inverted LCR can not only facilitate the formation of the non-allelic homologous recombination-mediated inversion polymorphism but it also promotes the occurrence of more complex rearrangements that can be associated with a large inversion, as well, but are mediated by a RBM. This further suggests that among the inversion polymorphism on 7p22.1, more complex rearrangements might be hidden. Furthermore, as the locus is embedded in a common fragile site (CFS) region, this rearrangement also supports the recently raised hypothesis that CFS sequence motifs may facilitate replication-based rearrangement mechanisms.
Similar content being viewed by others

Introduction
Constitutional mismatch repair deficiency (CMMRD) syndrome (MIM #276300) is an autosomal recessively inherited childhood cancer susceptibility syndrome caused by biallelic, that is, homozygous or compound heterozygous, germline mutations in one of the four DNA mismatch repair (MMR) genes, that is, MLH1 (MIM *120436), MSH2 (MIM *609309), MSH6 (MIM *600678) and PMS2 (MIM *600259).1 Individuals with CMMRD syndrome are at high risk to develop a diverse spectrum of malignancies already in childhood and adolescence.2 The spectrum mainly includes (i) T-cell non-Hodgkin's lymphoma and other haematological malignancies, (ii) high-grade gliomas and other brain/central nervous system tumours and (iii) colorectal and other cancers associated with Lynch syndrome (MIM #120435). In addition, a variety of other malignancies can also be CMMRD syndrome associated. Alterations of skin pigmentation, in particular multiple café-au-lait macules (CALM) reminiscent of neurofibromatosis type 1 (NF1), are frequently found in individuals with CMMRD syndrome. Besides pigmentation alterations, a set of other (non-)neoplastic features serve as diagnostic criteria for the suspected diagnosis of CMMRD in paediatric cancer patients according to a three-point scoring system developed by the European consortium ‘care for CMMRD’ (C4CMMRD).3
Approximately 60% of the so far reported individuals with CMMRD syndrome carry biallelic PMS2 germline mutations. This MMR gene is embedded within a complex genomic locus on chromosome 7 (band p22.1). The 3′-region of PMS2 (exons 9–15) is located in the telomeric copy of a 100-kb low-copy repeat (LCR) element (according to GRCh37; hg19), which is duplicated in inverse orientation. The centromeric copy of this LCR contains PMS2CL (according to GRCh37; hg19), a transcribed but non-functional PMS2 pseudogene (Supplementary Figure S1A).4 As regions containing LCRs are prone to recombination events, non-allelic homologous recombination (NAHR) between the LCR resolved through cross-over or gene conversion is believed to be the underlying cause of considerable sequence homogenisation particularly in a large central part of the duplicon. In this region, the two copies of the LCR cannot be distinguished from each other on the basis of the human genome reference sequence (GRCh37; hg19).5 Recombination-based sequence exchanges also affected the paralogs PMS2 and PMS2CL, situated at the inner ends of the LCR, which led to mainly functional hybrid PMS2 alleles that contain PMS2CL-derived sequences (as defined by NCBI RefSeq NC_000007.13), as well as hybrid PMS2CL alleles, with sequences derived from PMS2 (according to NCBI RefSeq NM_000535.5).5, 6, 7 The high prevalence of these hybrid alleles complicates mutation analysis, because in the region of frequent sequence exchange, that is, downstream of PMS2 exon 12, gene and pseudogene cannot be reliably distinguished on the basis of sequence differences with respect to their NCBI RefSeqs. Recombination-based sequence exchanges between the paralogs are thought to be a still on-going mechanism, which is also responsible for the generation of deleterious PMS2 alleles.8, 9 Furthermore, a known inversion polymorphism of ~0.7 Mb (Supplementary Figure S1B) is thought to result from intrachromosomal NAHR with cross-over between the two copies of the LCR.10, 11
A close proximity of the LCRs in the three dimensional space, which is a prerequisite for recombination to take place, might also bring replication forks together12 and thereby facilitate template switching events that may create other chromosomal rearrangements. Indeed, LCRs not only mediate recombination, but the nearby presence of LCRs is also one of the hallmarks of rearrangements thought to result from RBM such as fork stalling and template switching (FoSTeS) or microhomology-mediated break-induced replication (MMBIR).13, 14 According to the FoSTeS/MMBIR model, the active replication fork can stall during DNA replication and switch templates using complementary template microhomology to anneal and re-prime DNA replication.13, 14, 15, 16, 17, 18 Typically, the result is a deletion, if the invaded fork is located downstream (forward invasion), whereas switching to a fork located upstream (backward invasion) usually leads to a duplication. Depending on whether the lagging or leading strand in the new fork was used as replication template, the erroneously incorporated fragment would be in direct or inverted orientation with respect to its source locus.19 The FoSTeS/MMBIR model is highly apt to explain complex rearrangements with (multiple) deletions, duplications and inversions by postulating multiple template-switching events.18, 20, 21 Up to now, complex PMS2 alterations thought to result from RBM have not been described, but analysis of rearrangements in other genes has shown that RBMs are probably still under-recognised as molecular mechanism underlying (mainly complex) genomic, genic and exonic rearrangements.18, 22
Here, we describe a chromosomal rearrangement inactivating the PMS2 gene in an individual with CMMRD syndrome. Thorough characterisation of this complex rearrangement involving sequences of the inverted 100-kb LCR and a human endogenous retrovirus (HERV) element on 7p22.1 renders strong evidence that it results from a RBM.
Subjects and methods
Subjects
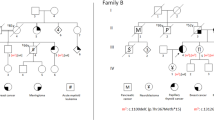
The index patient is a 5-year-old male. He is the third child of second-degree cousins of North-African ancestry (Figure 1, V3). All family members or their legal guardians who participated in this study gave written informed consent for genetic testing.

Pedigree of the family investigated. Roman numerals, generation identifier; circles, females; squares, males; unfilled symbols, unaffected relatives; filled symbols, relatives with cancer, that is, colon cancer (filled upper right quarter), lymphoma (filled lower right quarter) and brain tumour (filled lower left quarter); numbers below symbols, individual identifiers; numbers in symbols, number of offsprings.
PMS2 genetic analysis in the index patient
Sequencing of PMS2 RNA and genomic DNA
For direct cDNA sequencing of PMS2 according to the protocols developed in our laboratory,23 RNA was isolated from a short-term culture of the patient’s lymphocytes, which had been treated with puromycin before harvest to prevent nonsense-mediated decay (NMR)24 and was reverse transcribed using the SuperScript III Reverse Transcriptase (Invitrogen, Vienna, Austria). Using the resulting cDNA, RT-PCR products containing PMS2 exons 1–11, exons 10–15, exons 11–15, exons 1–9 and exons 7–12 were amplified with primers listed in Supplementary Table S1.
PMS2 exons 10 and 11 were amplified from genomic DNA with gene-specific primers published by Hendriks et al25 and van der Klift et al.7
All sequencing reactions were performed with Big Dye Terminator chemistry V1.1 (Applied Biosystems, Vienna, Austria) using the Applied Biosystems 3130 Genetic Analyzer for analyses (Applied Biosystems). Before sequencing, PCR products were treated with ExoSAP-IT (GE Healthcare, Vienna, Austria), and sequences were analysed with the Sequence Pilot algorithm, Version 4.1.2 Build 512 (JSI Medical Systems, Kippenheim, Germany).
The PMS2 variant and associated phenotype was submitted to the LOVD database (http://databases.lovd.nl/shared/variants/0000097022; Variant ID: 0000097022). Variants are named according to HGVS nomenclature using the reference transcript sequence NM_000535.5 for PMS2, NM_198097.3 for CCZ1B and NM_015622.5 for CCZ1 with the A of the start codon ATG being at position c.1. Exon numbering refers to NG_008466.1 for PMS2 and NC_000007.14 for CCZ1B/CCZ1.
PMS2-MLPA
The PMS2-MLPA kit P008-B1 (MRC-Holland, Amsterdam, The Netherlands) was used according to the manufacturer’s instructions. A total of 50–100 ng of gDNA was used per MLPA reaction. Each MLPA run included five appropriate reference DNA samples with an equal 2:2 distribution of PMS2 gene- and PMS2CL pseudogene-derived sequences in exon 13–15.9 MLPA results were analysed with the Sequence Pilot algorithm, Version 4.1.2 Build 512 (JSI Medical Systems).
Subtelomeric fluorescence in situ hybridisation
Subtelomeric fluorescence in situ hybridisation (FISH) was performed with the ToTelVysion Multi-Color FISH Probe set (Abbott, Wiesbaden, Germany) according to the manufacturer’s instructions.
GenomeWalker experiment to identify the rearrangement’s join point including the 3′-region of PMS2
For that purpose, 2.5 μg genomic DNA of the patient was blunt-end restriction digested using DraI and adaptors were ligated to the resulting DNA fragments. Nested PCR was performed with region‐specific primers (GSP1 and GSP2), both located within the non‐deleted sequence in PMS2 exon 11 (as confirmed by MLPA), and primers (AP1 and AP2), which hybridised to the ligated adaptors (primers are listed in Supplementary Table S2). Both PCRs were performed with the Advantage 2 PCR Kit (Clontech, Saint‐Germain‐en‐Laye, France), according to the manufacturer’s instructions.
3′-RACE PCR experiment to identify the rearrangement’s join point including the 5′-region of PMS2
For this experiment, it was pivotal to use 1 μg total RNA of the patient that was isolated from a phytohaemagglutinin-stimulated short-term lymphocyte culture treated with puromycine before cell harvest to prevent NMD.24 First strand cDNA synthesis was performed with the primer 3′-CDS according to the manufacturer’s instructions (SMARTer RACE 5′/3′ kit, Clontech). The 3′-RACE PCR was then performed with the Expand LongTemplate PCR kit (Roche, Vienna, Austria) according to the manufacturer’s instructions using the region‐specific primer RACE1 located in PMS2 exon 8 and the UPM primer (Supplementary Table S3). The 3′-RACE PCR product was purified (DNA Clean&Concentrator Kit, Zymo Research, CA, USA) and cloned (In-Fusion HD Cloning Kit, Clontech) before sequence analysis.
Array analysis for genomic copy number profiling
The Affymetrix CytoScan HD array platform (Affymetrix, Santa Clara, CA, USA) was used for genomic copy number profiling according to the manufacturer’s instructions. Probe allocation was based on the February 2009 human reference sequence (NCBI Build 37.1). Analysis was performed by the Helmholtz Centre, Munich.
Results
Clinical evaluation
A somnolent 5-year-old male was referred to the Department of Pediatric Hematology/Oncology at Hannover Medical School with symptoms of high intracranial pressure due to a brain tumour. Cranial computed tomography and magnetic resonance imaging were consistent with multifocal glioblastoma multiforme with progressive cerebral oedema and brain herniation through the foramen magnum. The boy (Figure 1; V3) is one of the six children born to a consanguine healthy couple of North African ancestry (Figure 1; IV1 and IV2; second-degree cousins). With regard to the family history, the parents and referring physicians reported that the paternal grandmother (Figure 1; III2) was diagnosed with colorectal cancer at 50 years of age and two older siblings (Figure 1; V1 and V2) died from non-further specified lymphomas at 4 and 6 years of age, respectively. Moreover, it was reported that two maternal cousins of the patient (Figure 1; V7 and V8) also died due to a lymphoma and a brain tumour at 9 and 12 years of age, respectively. Clinical examination of the boy displayed multiple CALM, which were also reported for the siblings already deceased, but were not present in the parents and the 4- and 8-year-old healthy siblings (Figure 1; V5 and V6).
With the glioblastoma multiforme (2 points), ≥2 CALM (2 points), the colorectal carcinoma of the paternal grandmother at the age of 50 (1 point), two of four siblings deceased due to childhood malignancies, most probably non-Hodgkin's lymphomas (2 times at least 1 point) and consanguineous parents (1 point), this patient reached at least eight points of the scoring system according to which any childhood cancer patient reaching at least three points should be suspected to have CMMRD syndrome.3 At the time the index patient was referred to the hospital, he was already in a terminal stage. In view of the palliative setting, the parents denied diagnostic biopsies. Therefore, no tissue was available for immunohistochemical staining, which usually directs mutation analysis to one of the four MMR genes.
PMS2 genetic analysis
Given that PMS2 is the most frequently affected MMR gene in CMMRD patients, we directly initiated PMS2 analysis starting with direct cDNA sequencing26 and refined MLPA analysis.9 Both primer pairs used to amplify the entire coding sequence of the PMS2 gene in two overlapping RT-PCR fragments (Supplementary Table S1) failed to generate a PCR product from cDNA of the patient (Supplementary Figure S2A). MLPA analysis using SALSA MLPA kit P008 (MRC-Holland) showed amplification loss of the first, that is, the most 5′, of three MLPA probes hybridising to PMS2 exon 11 (probe-no.: 14452-L00900, ie, E11-P1 in Supplementary Figure S3A). This MLPA result did not readily explain the failure to amplify cDNA with our standard primers. It was rather compatible with a homozygous deletion leading to loss of the 5′-region of PMS2 exon 11. The 3′-breakpoint of this suspected deletion would be located between the ligation site of the first and second MLPA probe in exon 11, which are only 77 nucleotides apart (E11-P1 and E11-P2 in Supplementary Figure S4A). To uncover the underlying genetic cause of the amplification loss of the MLPA probe, the entire PMS2 exon 11 and flanking intronic sequences were PCR amplified from genomic DNA of the patient with three gene-specific primer pairs7 (Supplementary Table S4). Notably, the sequence flanked by the first primer pair includes the ligation site of the first MLPA probe in exon 11 and the forward primer of the second primer pair (PMS2_11bf) binds to the same position as this MLPA probe (Supplementary Figure S4A). In agreement with the presence of a deletion including this MLPA probe binding site, these two primer pairs failed to generate a PCR product. The third primer pair readily amplified the 3′-region of exon 11 (Supplementary Figure S4A). To further define the extent of the deletion, PCR was performed with a presumably deletion-spanning primer pair (Supplementary Table S5). The forward primer of this primer pair was located in exon 10, which is specific for the PMS2 gene, as it is absent in PMS2CL. According to the MLPA results (Supplementary Figure S3A) and a PCR product including this exon and flanking intronic regions (Supplementary Table S4; Supplementary Figure S4B), exon 10 was present in two copies in the index patient. The reverse primer of the presumably deletion-spanning primer pair was located 3′ of the ligation site of the second MLPA probe (E11-P2) in exon 11, which was also present in two copies in the patient. However, this primer pair consistently failed to amplify any PCR product from the patient’s DNA (Supplementary Figure S4C), whereas the wild-type fragment of ~2.7 kb was readily amplified from a control DNA under all PCR conditions applied. Taking also into account that our RT-PCR experiments using primer pairs spanning the exon 10–11 transition failed to amplify PMS2 cDNA of the index patient, these results indicated a genomic rearrangement separating the 5'-part (exons 1–10) from the 3'-part (exons 11–15) of the gene. This assumption was further supported by RT-PCR experiments showing that the 5′-part of the gene (at least exons 1–9) were expressed, whereas no RT-PCR product could be generated with primers located in the 3′-part (exons 11–15) of the gene (Supplementary Figure S2B). A normal chromosomal karyotype in both parents, who were shown by MLPA analysis to be heterozygous carriers of the presumably alteration-associated deletion (Supplementary Figure S3B and C), rendered a translocation, involving the PMS2 locus, as underlying chromosomal alteration unlikely. This assumption was further supported by a regular subtelomeric FISH result in the mother (ToTelVysion Multi-Color FISH Probe set). Thus, an inversion, which separates the 5′-part from the 3′-part of the gene, associated with a small deletion was the most likely rearrangement.
Characterisation of the genomic rearrangement inactivating PMS2
In order to characterize the breakpoints of the assumed inversion, we first performed a GenomeWalker experiment aimed to identify the unknown sequence transferred to the 3′-part (exons 11–15) of PMS2. For that purpose, two gene-specific primers (GSP1 and GSP2) located within the non-deleted sequence in PMS2 exon 11 (as confirmed by MLPA) were combined in a nested PCR with the adaptor-specific primers AP1 and AP2. Sequencing into the 1.3-kb PCR product amplified with the nested primers GSP2 and AP2 indicated the transition of PMS2 exon 11 into the sequence of a HERV element originally located ~918 kb centromeric to the PMS2 gene (Supplementary Figure S5A). The orientation of the joined PMS2 exon 11 and HERV element sequences were in agreement with an inversion and contained a 2-bp (AG) microhomology (JP1 sequence in Figure 2a).

Possible FoSTeS/MMBIR-mediated rearrangement. The two characterised join points (JP) are shown in (a). A 2-bp microhomology (red letters) was observed at JP1 and a single-nucleotide change (turquoise letter in black box) was observed within the join point flanking region of JP2. SNPs are highlighted in green. (b) A possible FoSTeS/MMBIR-mediated rearrangement is depicted, which results from a first template switch (dotted arrow 1) from PMS2 (red bar) in the telomeric copy of the inverted LCR (light blue dashed box) to the HERV element (green bar), subsequent replication in reverse orientation until a second fork stalling in PMS2, followed by another template switch (dotted arrow 2) into intron 14 of the CCZ1B gene (purple bar) in the centromeric duplicon of the inverted LCR (dark blue dashed box). (c) Schematic representation of the rearranged allele. The red box indicates the region, which is duplicated as confirmed by array analysis. The deletion of the first 222 bp of PMS2 exon 11 is indicated. The non-transcribed PMS2 exon 11–15, as well as the transcribed fusion transcript of PMS2 exons 1–10 and a cryptic exon within CCZ1B intron 14, are shown as black boxes below the schematic representation of the rearranged allele. Exon numbering refers to NG_008466.1 (PMS2) and NC_000007.14 (CCZ1B).
Under the assumption of a simple inversion, different primer pairs were designed to amplify across the second breakpoint involving the 5′-part (exons 1–10) of PMS2 and the centromeric region of this HERV element (Supplementary Figure S5B). However, all primer pairs (Supplementary Table S6) consistently failed to amplify a breakpoint-spanning PCR product from the patient’s DNA. Therefore, the suspicion of a more complex chromosomal rearrangement was raised. In order to solve this complexity, we aimed at amplifying, now, into the unknown sequence, starting from the 5′-part of PMS2. For this purpose, we decided to perform rapid amplification of 3′-cDNA ends (3′-RACE). This experiment revealed a fusion transcript with a transition from PMS2 exon 10 into a sequence derived from intron 14 of either the CCZ1B gene or its paralog CCZ1, which have nearly identical sequences in this region, and are located in the centromeric and telomeric copy of the 100-kb inverted LCR, respectively (according to GRCh37; hg19) (Supplementary Figure S1A). Based on these results on RNA level, a primer pair was designed to span the breakpoint at gDNA level. The forward primer was located in PMS2 exon 10 and the reverse primer in the sequence of CCZ1B (or CCZ1) intron 14 that was present in the fusion transcript (Supplementary Table S7). Sequence analysis of the resulting PCR product showed that the breakpoint in PMS2 intron 10 is located at position c.1145–1 (ch7.hg19: g.6,027,252) and in CCZ1B intron 14 at position c.1393+595 (ch7.hg19: g.6,839,984) or CCZ1 intron 14 at position c.1393+597 (ch7.hg19: g.5,964,190). Again, the sequences were joined in an orientation indicating an inversion (according to the reference genome GRCh37; hg19). The join point of PMS2 intron 10 and CCZ1B (or CCZ1) intron 14 contained no microhomology but a single-nucleotide change (SNC) in the join point flanking region (see turquoise letter in black box in the JP2 sequence, Figure 2a). In contrast to SNPs (see green letter in JP2 sequence), SNCs are nucleotides that differ from the reference sequence of the human genome (hg19), but do not represent polymorphisms, as they are not listed in dbSNP. They are reported to be the result of error prone (repair) mechanisms.
Looking at each of the, here, characterised join points for itself, they could be mediated by non-replicative as well as RBM. Non-homologous end joining (NHEJ) is a repair mechanism, which is frequently associated with short microhomologies at the join points although not dependent upon it. Hence, NHEJ would be compatible with the 2-bp microhomology, which we observed in the JP1 sequence. Equally, the SNC observed at the second join point (JP2) could be the result of error prone NHEJ. However, microhomologies have also been observed at the breakpoints of genomic rearrangements mediated by a RBM such as FoSTeS or MMBIR14, 21, 27, 28 and the SNC could be the result of low-fidelity DNA polymerases or a replisome with reduced fidelity used in RBM.27 Taking also into account (i) that the sequences involved in the two identified join points suggested the presence of an inversion and (ii) that the MLPA results indicated a small deletion including the 5′-region of PMS2 exon 11, we expected a rather complex rearrangement. In comparison to NHEJ, which is a mechanism that explains primarily simple deletions with breakpoints in non-homologous sequences, RBM is more apt to explain complex rearrangements. Hence, assuming a FoSTeS/MMBIR mechanism and taking only the two identified join points for template switches into account, we envisioned three possible rearrangements (see the 'Discussion' section), which all included in addition to the observed features of the rearrangement a duplication of the region between CCZ1B (CCZ1) and the HERV element (ch7.hg19: g.6,839,984-6,945,161). This expected ~105-kb duplication was indeed confirmed by genome wide array analysis (Affymetrix CytoScan HD array platform; Supplementary Figure S6).
Discussion
Taken together we have identified several components of a complex rearrangement affecting PMS2. The most parsimonious explanation for the occurrence of this complex rearrangement is a RBM.
Three possible chromosomal rearrangements resulting from the two characterised template switches can be envisioned (Figure 2,Supplementary Figures S7 and S8). The possible rearrangements shown in Figure 2 and Supplementary Figure S7 occurred both on a source allele that corresponds to the reference sequence GRCh37 (hg19) with respect to the orientation of the sequence intervening the 100-kb LCR and the location of PMS2 in the telomeric copy of the inverted duplicon (Supplementary Figure S1A). For the rearrangement shown in Figure 2b, we propose a first fork stalling with a long-distance template switch from PMS2 exon 11 (ch7.hg19: g.6,027,029) to the HERV element located ~918 kb centromeric to PMS2 (ch7.hg19: g.6,945,161), subsequent replication in reverse orientation resulting in the inversion of almost the entire intervening sequence, then a second fork stalling with long-distance (~812 kb) template switch from PMS2 intron 10 (ch7.hg19: g.6,027,252) to the centromeric copy of the inverted 100-kb duplicon (CCZ1B intron 14; ch7.hg19: g.6,839,984). Resumption of the replication there resulted in a duplication of ~105 kb (ch7.hg19: g.6,839,984-6,945,161; red box in Figure 2c). This possible chromosomal rearrangement is deposited according to the HGVS nomenclature as ch7.hg19: g.[6027030_6027251del; 6027252_6945161inv; 6839984_6945161dup] in the LOVD/shared database. Of note, the rearranged allele shown in Figure 2c leads to an inversion of the entire sequence replicated between the first and the second template switch. This inversion resembles the known inversion polymorphism which affects the ~0.7-Mb sequence between the LCR (see Supplementary Figure S1B). The hybridisation probes used to characterize the inversion polymorphism by FISH analysis10, 11 would not be able to distinguish between the inversion of the, here, described complex rearrangement and an inversion of only the ~0.7-Mb sequence intervening the LCR on 7p22.1. Therefore, our data allow for the speculation that also other complex rearrangements could be concealed behind genomic inversions on 7p22.1. Moreover, our findings indicate that inversions on chromosome 7p22.1, which are thought to mainly origin from NAHR with cross-over between the highly homologous (>99%) LCRs, may also result from RBM.
A different rearrangement is generated from the same source allele if the two FoSTeS events occurred in inverse consecution: first switch from PMS2 intron 10 to CCZ1B intron 14, followed by replication until a second template switch from the HERV element to PMS2 exon 11 (Supplementary Figure S7). This rearrangement is not associated with an inversion of the ~0.7-Mb sequence intervening the LCR. The third possible rearrangement that can be envisioned (Supplementary Figure S8) would be created if the source allele would already harbour the described inversion polymorphism on 7p22.1 with the PMS2 and CCZ1 gene at the centromeric and the PMS2CL pseudogene, as well as the CCZ1B gene at the telomeric LCR and an inversion of the ~0.7-Mb sequence intervening the duplicons. In this case, FISH analysis would detect the (‘pre-existing’) inversion polymorphism.
The three, here, described possible rearrangements are the most parsimonious ones resulting from only two FoSTeS events and all three are compatible with the observed net result of deleted and duplicated sequences. The deletion in PMS2 exon 11 (ch7.hg19: g. 6,027,030_6,027,251del; c.1145_1367del) initially detected by MLPA results from the 222 bp of non-replicated sequence between the two FoSTeS events in PMS2. The position and orientation of the ~105-kb (ch7.hg19: g.6,839,984-6,945,161) duplication within the complex chromosomal rearrangement would be different in the three possible rearrangements (see the region highlighted by a red box in Figure 2c,Supplementary Figures S7B and S8B). Due to the large inverted regions (several hundred kb) and the high-sequence homology between the LCRs, it is not possible to further distinguish between the three postulated rearrangements and hence, define exactly which rearrangement is present in the index patient.
Of note, each of the here characterised FoSTeS/MMBIR events involved at least one sequence located within one of the two copies of the inverted 100-kb LCR showing >99% sequence homology. Hence, our findings might indicate that the particular chromosomal structure of the PMS2 locus predisposes this region not only to recombination events but also rearrangements resulting from RBM. LCR-mediated recombination implies that the LCRs, although separated by ~0.7 Mb, might be in close proximity in the three dimensional space. This close spatial proximity might also bring replication forks together,12 thereby facilitating the template switching that occurs in RBM. In line with this, the nearby presence of complex LCRs was initially reported to be one of the hallmarks of FoSTeS/MMBIR.13, 14
In addition, the chromosomal band 7p22.1 overlaps with a so-called common fragile site (CFS) termed FRA7B.29 CFS are seen in all individuals and, therefore, are a normal component of chromosome structure. Cytogenetic analyses originally identified at least 76 CFS which vary in size from 0.7–24.8 Mb.30, 31 CFS display a number of characteristics of unstable and highly recombinogenic DNA and are reported to break under replication stress.32 Among other reasons, DNA secondary structures33, 34, 35 and replication/transcription interference may impede replication fork progression directly.36, 37 Of note, one of the breakpoints in the, here, described rearrangement was located at the PMS2 intron 10 to exon 11 boundary. Given that transcription and splicing is tightly associated, this might render evidence that such interference could be at play. Furthermore, paucity of replication origins might force travelling forks to collapse.38 The stalled replication forks in turn result in unreplicated DNA,39 which may then evolve to DNA breaks—one of the main source of rearrangements.38 Re-initiation of replication from a stalled or collapsed fork is thought to usually involve homologous recombination.40 However, also FoSTeS/MMBIR was hypothesised to be a possible mechanism underlying the rearrangements at CFS,41 as FoSTeS events have the tendency to occur in regions, in which complex genomic architecture is thought to hamper replication fork progression. This was shown, for example, in a disease-causing complex rearrangement within the DMD gene.41 In support of this notion, we here describe another example of a complex chromosomal rearrangement mediated by FoSTeS/MMBIR at a CFS.
Conclusion
The here uncovered complex chromosomal rearrangement represents a novel mutational mechanism in the spectrum of PMS2 mutations. Two features of this rearrangement disrupting the PMS2 gene facilitated its identification. First, the rearrangement-associated deletion included exonic sequences that were readily detected to be deleted by MLPA analysis. Second, the rearrangement was homozygously present in a CMMRD patient. The latter was a prerequisite to easily notice the lack of transcripts containing the 3′-region of the PMS2 gene (exons 11–15) by direct cDNA sequencing. The fact that the results of MLPA analysis suggesting a simple intragenic deletion could not readily explain the results of direct cDNA sequencing prompted us to further characterize the underlying genetic alteration. It is well conceivable that Lynch and CMMRD syndrome patients with so far unidentified PMS2 mutations carry inversion-associated or other complex rearrangements that escape gDNA-based mutation analysis protocols. This was shown, for example, by the identification of an SVA insertion in PMS2 intron 7, which was resolved only by RNA-based mutation analysis.42 Of note, gross deletions affecting one or more exons of the PMS2 gene represent 17–33% of the disease-causing PMS2 mutations found in Lynch or CMMRD syndrome patients.9 The mutation database HGMD Professional (Spring 2015.4 release) lists 201 different disease-causing PMS2 mutations of which 65 (32%) are gross intragenic deletions. So far, only 10 of these have been characterised at molecular level.7, 9, 43, 44 Analyses of the breakpoints of these deletions show that all of them involve Alu elements suggesting that they were mainly generated by NAHR (Supplementary Table S8). Still, as a recent study on the genetic mechanisms responsible for intragenic gross deletions inactivating the NF1 gene suggests that FoSTeS/MMBIR is an underestimated cause of this type of gene alterations,22 it may be worthwhile to test whether also RBM may have a role for the high frequency of non-recurrent (intragenic) deletions affecting the PMS2 gene.
References
Wimmer K, Kratz CP : Constitutional mismatch repair-deficiency syndrome. Haematologica 2010; 95: 699–701.
Vasen H, Ghorbanoghli Z, Bourdeaut F et al: Guidelines for surveillance of individuals with constitutional mismatch repair-deficiency proposed by the European Consortium ‘Care for CMMR-D’(C4CMMR-D). J Med Genet 2014; 51: 283–293.
Wimmer K, Kratz CP, Vasen HF et al: Diagnostic criteria for constitutional mismatch repair deficiency syndrome: suggestions of the European consortium 'care for CMMRD' (C4CMMRD). J Med Genet 2014; 51: 355–365.
De Vos M, Hayward BE, Picton S, Sheridan E, Bonthron DT : Novel PMS2 pseudogenes can conceal recessive mutations causing a distinctive childhood cancer syndrome. Am J Hum Genet 2004; 74: 954–964.
Hayward BE, De Vos M, Valleley EM et al: Extensive gene conversion at the PMS2 DNA mismatch repair locus. Hum Mutat 2007; 28: 424–430.
Ganster C, Wernstedt A, Kehrer-Sawatzki H et al: Functional PMS2 hybrid alleles containing a pseudogene-specific missense variant trace back to a single ancient intrachromosomal recombination event. Hum Mutat 2010; 31: 552–560.
van der Klift HM, Tops CM, Bik EC et al: Quantification of sequence exchange events between PMS2 and PMS2CL provides a basis for improved mutation scanning of Lynch syndrome patients. Hum Mutat 2010; 31: 578–587.
Auclair J, Leroux D, Desseigne F et al: Novel biallelic mutations in MSH6 and PMS2 genes: gene conversion as a likely cause of PMS2 gene inactivation. Hum Mutat 2007; 28: 1084–1090.
Wernstedt A, Valtorta E, Armelao F et al: Improved multiplex ligation-dependent probe amplification analysis identifies a deleterious PMS2 allele generated by recombination with crossover between PMS2 and PMS2CL. Genes Chromosomes Cancer 2012; 51: 819–831.
Feuk L, MacDonald JR, Tang T et al: Discovery of human inversion polymorphisms by comparative analysis of human and chimpanzee DNA sequence assemblies. PLoS Genet 2005; 1: e56.
Szamalek JM, Cooper DN, Schempp W et al: Polymorphic micro-inversions contribute to the genomic variability of humans and chimpanzees. Hum Genet 2006; 119: 103–112.
Beck CR, Carvalho CM, Banser L et al: Complex genomic rearrangements at the PLP1 locus include triplication and quadruplication. PLoS Genet 2015; 11: e1005050.
Lee JA, Carvalho CM, Lupski JR : A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell 2007; 131: 1235–1247.
Hastings P, Ira G, Lupski JR : A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet 2009; 5: e1000327.
Liu P, Carvalho CM, Hastings P, Lupski JR : Mechanisms for recurrent and complex human genomic rearrangements. Curr Opin Genet Dev 2012; 22: 211–220.
Hastings P, Lupski JR, Rosenberg SM, Ira G : Mechanisms of change in gene copy number. Nat Rev Genet 2009; 10: 551–564.
Arlt MF, Mulle JG, Schaibley VM et al: Replication stress induces genome-wide copy number changes in human cells that resemble polymorphic and pathogenic variants. Am J Hum Genet 2009; 84: 339–350.
Zhang F, Khajavi M, Connolly AM, Towne CF, Batish SD, Lupski JR : The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat Genet 2009; 41: 849–853.
Gu W, Zhang F, Lupski JR : Mechanisms for human genomic rearrangements. Pathogenetics 2008; 1: 4.
Carvalho CM, Ramocki MB, Pehlivan D et al: Inverted genomic segments and complex triplication rearrangements are mediated by inverted repeats in the human genome. Nat Genet 2011; 43: 1074–1081.
Zhang F, Seeman P, Liu P et al: Mechanisms for nonrecurrent genomic rearrangements associated with CMT1A or HNPP: rare CNVs as a cause for missing heritability. Am J Hum Genet 2010; 86: 892–903.
Hsiao MC, Piotrowski A, Callens T et al: Decoding NF1 intragenic copy-number changes. Am J Hum Genet 2015; 97: 238–249.
Etzler J, Peyrl A, Zatkova A et al: RNA-based mutation analysis identifies an unusual MSH6 splicing defect and circumvents PMS2 pseudogene interference. Hum Mutat 2008; 29: 299–305.
Andreutti-Zaugg C, Scott RJ, Iggo R : Inhibition of nonsense-mediated messenger RNA decay in clinical samples facilitates detection of human MSH2 mutations with an in vivo fusion protein assay and conventional techniques. Cancer Res 1997; 57: 3288–3293.
Hendriks YM, Jagmohan-Changur S, van der Klift HM et al: Heterozygous mutations in PMS2 cause hereditary nonpolyposis colorectal carcinoma (Lynch syndrome). Gastroenterology 2006; 130: 312–322.
Wimmer K, Wernstedt A : PMS2 gene mutational analysis: direct cDNA sequencing to circumvent pseudogene interference. Pseudogenes: Springer 2014; 1167: 289–302.
Carvalho CM, Pehlivan D, Ramocki MB et al: Replicative mechanisms for CNV formation are error prone. Nat Genet 2013; 45: 1319–1326.
Carvalho CM, Zhang F, Liu P et al: Complex rearrangements in patients with duplications of MECP2 can occur by fork stalling and template switching. Hum Mol Genet 2009; 18: 2188–2203.
Bosco N, Pelliccia F, Rocchi A : Characterization of FRA7B, a human common fragile site mapped at the 7p chromosome terminal region. Cancer Genet Cytogenet 2010; 202: 47–52.
Lukusa T, Fryns J-P : Human chromosome fragility. Biochim Biophys Acta 2008; 1779: 3–16.
Mrasek K, Schoder C, Teichmann A-C et al: Global screening and extended nomenclature for 230 aphidicolin-inducible fragile sites, including 61 yet unreported ones. Int J Oncol 2010; 36: 929–940.
Arlt M, Casper A, Glover T : Common fragile sites. Cytogenet Genome Res 2003; 100: 92–100.
Arlt MF, Durkin SG, Ragland RL, Glover TW : Common fragile sites as targets for chromosome rearrangements. DNA Repair 2006; 5: 1126–1135.
Zlotorynski E, Rahat A, Skaug J et al: Molecular basis for expression of common and rare fragile sites. Mol Cell Biol 2003; 23: 7143–7151.
Dillon LW, Burrow AA, Wang Y-H : DNA instability at chromosomal fragile sites in cancer. Curr Genomics 2010; 11: 326.
Aguilera A, García-Muse T : R loops: from transcription byproducts to threats to genome stability. Mol Cell 2012; 46: 115–124.
Helmrich A, Ballarino M, Tora L : Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol Cell 2011; 44: 966–977.
Le Tallec B, Koundrioukoff S, Wilhelm T, Letessier A, Brison O, Debatisse M : Updating the mechanisms of common fragile site instability: how to reconcile the different views? Cell Mol Life Sci 2014; 71: 4489–4494.
Casper AM, Nghiem P, Arlt MF, Glover TW : ATR regulates fragile site stability. Cell 2002; 111: 779–789.
Rothkamm K, Krüger I, Thompson LH, Löbrich M : Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol Cell Biol 2003; 23: 5706–5715.
Mitsui J, Takahashi Y, Goto J et al: Mechanisms of genomic instabilities underlying two common fragile-site-associated loci, PARK2 and DMD, in germ cell and cancer cell lines. Am J Hum Genet 2010; 87: 75–89.
van der Klift HM, Tops CM, Hes FJ, Devilee P, Wijnen JT : Insertion of an SVA element, a nonautonomous retrotransposon, in PMS2 intron 7 as a novel cause of Lynch syndrome. Hum Mutat 2012; 33: 1051–1055.
Borràs E, Pineda M, Cadiñanos J et al: Refining the role of PMS2 in Lynch syndrome: germline mutational analysis improved by comprehensive assessment of variants. J Med Genet 2013; 50: 552–563.
Vaughn CP, Baker CL, Samowitz WS, Swensen JJ : The frequency of previously undetectable deletions involving 3′ exons of the PMS2 gene. Genes Chromosomes Cancer 2013; 52: 107–112.
Acknowledgements
We thank the technical staff of the Institute of Human Genetics in Innsbruck for excellent technical support and the Helmholtz Institute, Munich, Germany for performing the CytoScan HD analysis.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Vogt, J., Wernstedt, A., Ripperger, T. et al. PMS2 inactivation by a complex rearrangement involving an HERV retroelement and the inverted 100-kb duplicon on 7p22.1. Eur J Hum Genet 24, 1598–1604 (2016). https://doi.org/10.1038/ejhg.2016.75
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2016.75
This article is cited by
-
Detecting inversions in routine molecular diagnosis in MMR genes
Familial Cancer (2022)
-
Seltene Tumordispositionssyndrome mit Manifestation im Kindesalter
Medizinische Genetik (2017)
