
Abstract
Essential tremor (ET) is one of the most common causes of tremor in humans. Despite its high heritability and prevalence, few susceptibility genes for ET have been identified. To identify ET genes, whole-exome sequencing was performed in 37 early-onset ET families with an autosomal-dominant inheritance pattern. We identified candidate genes for follow-up functional studies in five ET families. In two independent families, we identified variants predicted to affect function in the nitric oxide (NO) synthase 3 gene (NOS3) that cosegregated with disease. NOS3 is highly expressed in the central nervous system (including cerebellum), neurons and endothelial cells, and is one of three enzymes that converts l-arginine to the neurotransmitter NO. In one family, a heterozygous variant, c.46G>A (p.(Gly16Ser)), in NOS3, was identified in three affected ET cases and was absent in an unaffected family member; and in a second family, a heterozygous variant, c.164C>T (p.(Pro55Leu)), was identified in three affected ET cases (dizygotic twins and their mother). Both variants result in amino-acid substitutions of highly conserved amino-acid residues that are predicted to be deleterious and damaging by in silico analysis. In three independent families, variants predicted to affect function were also identified in other genes, including KCNS2 (KV9.2), HAPLN4 (BRAL2) and USP46. These genes are highly expressed in the cerebellum and Purkinje cells, and influence function of the gamma-amino butyric acid (GABA)-ergic system. This is in concordance with recent evidence that the pathophysiological process in ET involves cerebellar dysfunction and possibly cerebellar degeneration with a reduction in Purkinje cells, and a decrease in GABA-ergic tone.
Similar content being viewed by others
Introduction
Essential tremor (ET) is a chronic, progressive neurological disease.1 Its hallmark motor feature is a 4–12-Hz kinetic tremor that occurs during voluntary movements such as writing, eating and drinking. At disease onset, the tremor not only affects the hands and arms but it may also eventually spread to involve the head (ie, neck), voice, jaw and other body regions.2 Given the presence of etiological, clinical, pharmacological response and pathological heterogeneity, there is growing support for the idea that ET may be a family of diseases whose central defining feature is kinetic tremor of the arms, and therefore it might more appropriately be called ‘the essential tremors’.3
Family studies4, 5 and twin studies6, 7 provide strong evidence for a genetic contribution to ET, with heritability estimates ranging from 45 to 90% in twin studies.6, 7 Despite this high heritability, the field of ET genetics has made only limited advances. Previously, we and others reported that ET aggregates in families, with many families containing multiple members with ET.8, 9, 10 Most studies indicate that at least 30–70% of ET patients have a family history, with the vast majority (>80%) of young onset (⩽40 years old) cases reporting ⩾1 affected first-degree relative.11 To date, only three published genome-wide linkage scans have been performed, all in North American or Icelandic ET families.12, 13, 14 These studies provided evidence for genetic loci harboring ET genes on chromosomes 3q13 (ETM1; OMIM: 190300),12 2p22-p25 (ETM2; OMIM: 602134)13 and 6p23 (ETM3; OMIM: 611456).14 Several studies have attempted to replicate linkage to ETM1,15, 16, 17 ETM215, 18, 19 and ETM3, without success (no LOD score >2.0), and the genes and causal variants for these loci (ETM1, ETM2 and ETM3) have yet to be identified. Recently, using a linkage and a whole-exome sequencing approach, the fused in sarcoma/translated in liposarcoma (FUS/TLS) gene (Chr16p11.2) was identified as a candidate gene in a large family with ET from Quebec.20 Subsequent studies,21, 22, 23 including our own, indicate that variants in FUS are an extremely rare or family-specific cause of ET, and without functional studies, the pathogenicity of variants identified so far ((c.868C>T, p.(Q290*)), RefSeq accession number NM_004960.320 and c.1129C>T p.(R377W), RefSeq accession number NM_004960.3 reported in 1 patient with family history of ET24 is unknown.
Despite its remarkably high prevalence, the pathophysiology of ET is still poorly understood and current debate as to whether it is a functional or neurodegenerative disease.25, 26 There is considerable evidence from clinical, neuroimaging and physiological studies, of cerebellar involvement,27, 28 and an emerging literature that documents a variety of changes, some of which are degenerative, in the Purkinje cell population.29, 30 There is also a related literature that indicates a possible change in brain gamma-amino butyric acid (GABA) tone in ET.31, 32
Materials and methods
Study participants and clinical diagnosis
To identify genes for ET, we enrolled probands (affected with ET) and relatives in a family study of ET at Columbia University, NY, USA (2011–2014). The study was approved by the Institutional Review Board at Columbia University and written informed consent was obtained from all enrollees. The criteria for enrollment were as follows: (1) the proband had early-onset ET with age at onset ⩽50 years; (2) the proband had a diagnoses of definite or probable ET; (3) in addition to the proband there were at least two affected relatives in the family; (4) additional affected and unaffected family members were willing to participate in the study; and (5) the families contained more than two affected individuals in different generations. For the genetic analyses, we excluded enrollees who had been diagnosed with Parkinson’s disease (PD) or dystonia. The final sample includes 52 families (52 probands (affected with ET)) and 155 relatives. The number of affected individuals enrolled per family ranged from 3 to 7 (mean=4.1). An in-person evaluation was performed on all participants, during which they completed demographic, medical history and family history questionnaires, and underwent a videotaped neurological examination, from which a total tremor score (range 0–36) was assigned.10, 33, 34, 35, 36, 37 After review of the questionnaires and videotaped examinations, the diagnosis of ET was then reconfirmed by a senior neurologist specializing in movement disorders (EDL) using reliable and valid research criteria.38 All ET diagnoses (possible, probable and definite) required, at a minimum, moderate or greater amplitude kinetic tremor on at least three tasks, and an absence of other etiologies (eg, dystonia, PD and drug-induced tremor). Probable ET required such tremor on at least four tasks and definite ET required this as well as postural tremor of at least moderate amplitude.38 As such, all ET diagnoses also met the requirements outlined in the Consensus Statement on Tremor of the Movement Disorders Society.39 At the time of starting the genetic analyses, 37 families had completed all clinical assessments and evaluations (videotape, diagnosis of ET by EDL and isolated DNA), and were selected for whole-exome sequencing.
An additional 95 unrelated ET cases enrolled in a clinical–epidemiological study at Columbia University21 were also screened by Sanger sequencing for KCNS2 coding variants.
Whole-exome sequencing analysis
Genomic DNA was isolated from peripheral blood cells using standard methods. Whole-exome sequencing was performed on the genomic DNA of at least two most distantly related affected (definite, probable or possible ET diagnosis) individuals from each of 37 total families. In some families, >2 affected individuals were sequenced. The pedigrees for 37 families, indicating that family members were exome sequenced, are shown in Supplementary Figure S1. All samples were processed using the Agilent SureSelect XT kit (Agilent Technologies, Santa Clara, CA, USA) for library preparation and exome captured using the All Exon v5+UTRs library (Agilent Technologies). Paired-end sequencing was performed at >40 × coverage per sample. Obtained libraries were sequenced on the Illumina HiSeq2500 instrument (Illumina, Inc., San Diego, CA, USA). Sequence alignment to the human reference genome (UCSC hg19) was performed using the Burrows–Wheeler Aligner algorithm,40 and variant calling was performed using the Genome Analysis Toolkit (Broad Institute, Cambridge, MA, USA).41 The following criteria were used to generate a list of variants for each family for follow-up genotyping of other family members and cosegregation analysis: (1) on the assumption that disease-causing variants were rare, we excluded all common single-nucleotide polymorphisms (MAF>1%) in dbSNP v.137; (2) the variant was shared between individuals with ET within a family; and (3) the variant was ranked by the software tool pVAAST (see Web Resources)42 (using a dominant model and the maximum number of permutations of 1 000 000) with P-value <0.05.
Genotyping
Follow-up genotyping was performed using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (Sequenom, San Diego, CA, USA) with Sequenom iPlex Gold custom assays designed using MassARRAY assay design software version 4.0 (Sequenom). Details of the variants that were genotyped are provided in Supplementary Table S1.
From the cosegregation analysis, we identified a list of annotated ‘candidate’ variants that were shared exclusively by affected ET patients within a family. Annotation of cosegregating candidate variants was performed at the gene and variant level (Supplementary Materials and Methods).
Availability of data
All phenotype, genotype and exome data will be released and deposited in the database of Genotypes and Phenotypes (dbGaP: http://www.ncbi.nlm.nih.gov/gap) of the National Center for Biotechnology Information. The study titled ‘Identification of Susceptibility Genes for Essential Tremor’ received the dbGaP accession phs000966.v1.p1.
Results
The demographic and clinical characteristics of affected ET patients and unaffected family members that were sequenced are shown in Table 1. The average age at onset of tremor in ET patients was 24.5 years (±17.2) and duration of tremor was 35.3 years (±19.5). Ten percent of ET patients reported Ashkenazi Jewish ancestry.
We identified variants predicted to affect function in five ET families (Table 2). Cosegregating variants were considered to be candidates based on the following criteria: (1) the variant occurred at a highly conserved nucleotide or amino-acid residue; (2) amino acid changes were predicted to be damaging to the protein by one or more prediction tools (provean, SIFT and mutation taster); (3) the gene is expressed in the cerebellum (including Purkinje, granule, stellate and basket cells); and (4) the gene function is implicated in the pathophysiological process in ET. In two independent families (families A and B), we identified variants in the nitric oxide (NO) synthase 3 gene (NOS3) that cosegregated with disease. In family A, a heterozygous variant, c.46G>A (p.(Gly16Ser)), located in NOS3, was present in all three affected ET cases and absent in an unaffected family member. In family B, a heterozygous variant, c.164C>T (p.(Pro55Leu)), was identified in three affected ET cases that included dizygotic twins and their mother. These NOS3 variants are absent from the 1000 genomes project (TGP) database but are present in the heterozygous state in the exome aggregation consortium (ExAC) data set with an allele frequency of 1.969 × 10−5 and 5.145 × 10−5. The father was also affected with ET but did not carry the heterozygous variant identified in other family members, and family history information was not available for him as he was adopted, suggesting the possibility of a phenocopy. NOS3 was ranked 32 (P-value 0.0133) in the pVAAST analysis (Supplementary Materials and Methods). Both variants result in amino-acid substitutions of highly conserved amino-acid residues that are predicted to be deleterious and damaging by in silico analysis (both mutations by SIFT and mutation taster). The main clinical features of six affected individuals from two families with NOS3 variants are shown in Table 3.
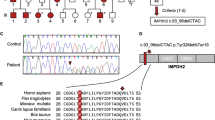
In family C, a heterozygous variant was identified in the gene KCNS2 (KV9.2), c.1137T>A, resulting in a missense amino-acid substitution (p.(Asp379Glu)) and ranked 52 (P-value 0.027) in the pVAAST analysis (Supplementary Materials and Methods). The variant is absent from the TGP and ExAC databases. This variant was present in all affected ET patients but was absent in an unaffected family member (Figure 1). KCNS2 was also identified as a candidate gene in a significant linkage interval (P2 LOD 3.312 and P1 LOD 2.729) on hg19 chr8: g.94960934_101969832 (Supplementary Materials and Methods, and Supplementary Table S2) by nonparametric linkage analysis in 37 ET families. Three in silico prediction programs, provean,43 SIFT44 and mutation taster,45 predict that this amino-acid substitution is intolerable, deleterious and damaging to the structure and function of the KCNS2 protein. Furthermore, the asparagine residue at position 379 is highly evolutionarily conserved, suggesting that this residue is essential for channel function (Figure 2). KCNS2 also shows high homology to Drosophila Shab, a K+ channel that regulates membrane excitability and synaptic transmission in many central nervous system (CNS) neurons, in addition to the neuromuscular junction, with 42% amino-acid identity and 63% amino-acid similarity. Protein alignment of Drosophila Shab with KCNS2 shows that the asparagine residue (and adjacent amino acids) at position 379 in KCNS2 is conserved in Shab (Figure 2). We also evaluated the effect of the variant on the protein structure (residue range 332–413) using Swiss-Pdb viewer (Deep View version 4.0, see Web Resources; Figure 3). The variant, c.1137T>A (p.(Asp379Glu)), alters the molecular surface, electrostatic potential and creates a strong hydrogen bond that is absent from the wild-type protein (Figure 3). We sequenced the coding regions of the KCNS2 gene in an additional 95 unrelated ET cases enrolled in a clinical–epidemiological study at Columbia University.21 We did not identify novel variants predicted to affect function in KCNS2, nor did we identify the KCNS2 variant, c.1137T>A (p.(Asp379Glu)), identified in family C, in any of the 95 ET cases analyzed.

Pedigrees with variants in the genes NOS3 (families a and b) KCNS2 (family c), HAPLN4 (family d) and USP46 (family e). Genetic pedigrees for families with likely pathogenic mutations are shown. The generation in each pedigree is indicated by roman numerals. The proband is indicated by an arrowhead. A ‘+’ symbol indicates subjects that were exome sequenced. Below each subject with DNA available for genetic analysis, the subject id (00X), age at tremor onset and date of birth are indicated. Symbol shading is as follows: definite ET, symbols completely black; probable ET, symbols half vertical black fill; possible ET, symbols with a quadrant in black; and unaffected, clear symbol.

Evolutionary conservation of KCNS2. Evolutionary conservation of KCNS2 at the protein level. The asparagine residue indicated in blue at position 379 in Human KCNS2 is highly conserved across species including chimpanzee, rhesus macaque, mouse and pufferfish. The non-synonymous substitution to glutamic acid identified in family 68 is indicated in red. KCNS2 also shows high homology to Drosophila Shab, a K+ with 42% amino-acid identity and 63% amino-acid similarity. Protein alignment of Drosophila Shab with KCNS2 shows that the asparagine residue (and adjacent amino acids) at position 379 in KCNS2 is conserved in Shab.

Model 3D protein structure of KCNS2. A model 3D structure of KCNS2 was generated using Swiss-Pdb viewer (Deep View version 4.0). The modeled residue range includes amino acids 332–413, which includes the KCNS2 variant c.1137T>A (p.(Asp379Glu)), identified in family C. The amino acid 379 is indicated. The variant alters the molecular surface, electrostatic interactions (indicated by colored dots) and creates a strong hydrogen bond (green dotted line).
In family D, we identified a heterozygous variant, c.1048G>C (p.(Gly350Arg)), in hyaluronan and proteoglycan link protein 4 (HAPLN4; pVAAST rank 38, P-value 0.0167) that was identified in three affected ET cases. DNA was not available for other family members that reside in the United Kingdom and were unwilling to participate. Three in silico prediction programs, provean,43 SIFT44 and mutation taster,45 predict that this amino-acid substitution is intolerable, deleterious and damaging to the structure and function of the HAPLN protein. Furthermore, the glycine residue at position 350 is highly evolutionarily conserved, suggesting that this residue is essential for function of the protein. The variant is absent from the TGP database and present in the ExAC database with an allele frequency of 0.00002917.
In family E, we identified a heterozygous variant, c.398C>T (p.(Ala133Val)) in ubiquitin-specific protease 46 (USP46), which cosegregated with ET in the family (pVAAST rank 5, P-value 0.00228). The variant was present in five ET cases. DNA was not available for testing from other family members. Although only one in silico prediction program (mutation taster) predicts that the amino-acid substitution in USP46 is disease causing, the alanine residue at position 133 is highly conserved in several species (chimpanzee, macaque, cat, mouse, chicken, pufferfish and zebrafish). The variant is absent from the TGP genomes and ExAC databases.
Discussion
Among the 37 early-onset ET families analyzed, we identified four promising candidate genes in five families. Functional studies will be needed to validate our findings and determine their mechanism of action (dominant gain-of-function or dominant negative) and role in ET.
In two families (A and B), we identified variants predicted to affect function in NOS3. NOS3, one of three enzymes that converts l-arginine into the neurotransmitter NO, is the major NO synthase (NOS) isoform expressed in endothelial cells and has an important role in vasculature homeostasis.46 NOS3 is also highly expressed in the CNS (including the cerebellum), neurons and endothelial cells.46 In the brain, NO mediates neuronal survival, synaptic plasticity, vascular smooth muscle relaxation and endothelial cell permeability. The cerebellum expresses high levels of NOS in granule, stellate and basket cells.47 The NOS pathway has been implicated in the pathogenesis of Alzheimer’s disease (AD), PD, and cerebrovascular disease and stroke, and previous studies have also demonstrated aberrant expression of the NOS3 gene in neurons, glial and endothelial cells in AD brains.46, 47, 48 The Glu/Glu genotype at p.(Asp298Glu) (rs1799983; ClinVar pathogenic allele) has been reported as a risk factor for AD in several case–control studies.
Recent findings from several studies, including our own, indicate that the pathophysiological process in ET involves the cerebellum. There is also some discussion as to whether this is associated with cerebellar degeneration as well as a decrease in GABA-ergic activity in deep cerebellar neurons, disinhibition in output to the deep cerebellar neurons and an increase in rhythmic activity of the thalamocortical circuit.25, 26 Significantly, in the current study, we identified variants in three genes in independent ET families, which include KCNS2 (KV9.2) (family C), HAPLN4 (BRAL2) (family D) and USP46 (family E), each of which is highly expressed in Purkinje cells.
KCNS2 (KV9.2), a K+ channel α-subunit that is highly and selectively expressed in the brain, modulates the activity of the KV2.1 and KV2.2 channels.49 A similar localization of expression of KCNS2 (KV9.2) and KV2.1 and KV2.2 has been observed in the Purkinje and granular cells in the cerebellum.49 KCNS2 also shows high homology to Drosophila Shab, a K+ channel that is widely expressed in the CNS and peripheral nervous systems, and that interacts with the Drosophila Shaker channel in the regulation of synaptic transmission.50 Indeed, these channels regulate membrane excitability and synaptic transmission in many central neurons and also in the neuromuscular junction.50 Drosophila Shaker mutants display leg shaking under anesthesia,51 abnormal spike bursting in motor circuits52 and greatly enhanced neurotransmission at neuromuscular junctions.53 A tremor phenotype has been described in patients with variants in related potassium channel family members (eg, KCNA1, KV1.1).54 Further, in other K+ channels mouse models (KV3.1 and KV3.3 K+ channel), mice display severe motor deficits, including tremor, myoclonus and ataxic gait.55, 56
In family D, the gene, brain link 2 (BRAL2; also known as HAPLN4) was identified as a candidate. It is of special interest that BRLA2 is highly expressed throughout the GABA-ergic neurons of the cerebellum, including Purkinje cells and basket neurons, both of which have been shown in some studies to be abnormal in ET57, 58 in postmortem studies of humans with ET. Furthermore, the Purkinje cells, and their entire GABA-ergic output, are directed at the neurons in the deep cerebellar nuclei. Therefore, a reduction in Purkinje cell output or a reduction in the receptivity of deep cerebellar nuclei to GABA could result in tremor.59 Of particular interest is that mice deficient for BRAL2 have attenuated perineuronal nets, altered localization of brevican and show a slight decrease in the number of synapses in deep cerebellar nuclei neurons. Interestingly, in a mouse model of a related link protein family member, BRAL1, CNS axonal nerve conduction is markedly decreased without changes in the clustering or transition of ion channels at the nodes or in the tissue morphology around the nodes of Ranvier. BRAL1 mice did however changes in the extracellular space diffusion parameters, suggesting a reduction in the diffusion hindrances in the white matter of mutant mice. Behavioral studies or other phenotypic observations such as tremor have not been described for the BRAL2-deficient mice, but further characterization of these mice in light of our findings is warranted.60
USP46 was identified as a candidate gene in family E. USP46 belongs to a family of deubiquitinating enzymes that regulate diverse cellular functions by cleaving ubiquitin from specific protein substrates. They have been implicated in a wide variety of biological processes and disease processes including cancer and neurodegenerative disease.61, 62, 63, 64 Usp46 is strongly expressed in different brain regions including the cerebellum. In a Usp46 mouse mutant model,65 defects in the GABA-ergic system were identified with a marked reduction in GAD67 expression. Because GAD67 catalyzes the decarboxylation of glutamate to GABA, Usp46 has been proposed to be involved in GABA synthesis. Thus, it is likely that Usp46 extensively affects the GABA-ergic system and controls a broad range of behavioral phenotypes.65, 66
One of the strengths of the study is the use of diagnostic criteria that are both reliable and valid, and that were designed specifically for genetic studies of ET, the Washington Heights Inwood Genetic Study of ET. All diagnoses were assigned by a senior movement disorder neurologist based on a detailed, videotaped neurological examination that contained a rigorous assessment of tremor and tremor phenomenology. All ET diagnoses required tremors during a minimum of three separate tasks, with diagnoses of probable and definite requiring tremor during four tasks. Another strength is our focus on families with an early age at ET onset, with a mean age at onset of 24.5 years (±17.2), which may have enriched the sample for disease susceptibility variants.
Although we have identified a number of interesting candidates, future functional studies are needed to evaluate the pathogenicity of the variants identified in these candidate genes. However, gene and variant level annotation suggest that the variants are predicted to be deleterious and damaging, and functional annotation suggests that many of the candidate genes are likely to have a role in ET pathogenesis. Follow-up studies in cellular and animal models will be needed to determine the role of these genes in ET.
References
Louis ED : Clinical practice. Essential tremor. N Engl J Med 2001; 345: 887–891.
Louis ED, Gerbin M, Galecki M : Essential tremor 10, 20, 30, 40: clinical snapshots of the disease by decade of duration. Eur J Neurol 2013; 20: 949–954.
Louis ED : 'Essential tremor' or 'the essential tremors': is this one disease or a family of diseases? Neuroepidemiology 2013; 42: 81–89.
Bain PG, Findley LJ, Thompson PD et al: A study of hereditary essential tremor. Brain 1994; 117: 805–824.
Findley LJ : Epidemiology and genetics of essential tremor. Neurology 2000; 54: S8–S13.
Tanner CM, Goldman SM, Lyons KE et al: Essential tremor in twins: an assessment of genetic vs environmental determinants of etiology. Neurology 2001; 57: 1389–1391.
Lorenz D, Frederiksen H, Moises H, Kopper K, Deuschl G, Christensen K : High concordance for essential tremor in monozygotic twins of old age. Neurology 2004; 62: 208–211.
Rautakorpi I, Takala J, Marttila RJ, Sievers K, Rinne UK : Essential tremor in a Finnish population. Acta Neurol Scand 1982; 66: 58–67.
Louis ED, Ford B, Frucht S, Rabinowitz D, Ottman R : Evidence for familial aggregation of tremor in normal individuals. Neurology 2001; 57: 110–114.
Louis ED, Ford B, Frucht S, Barnes LF, X-Tang M, Ottman R : Risk of tremor and impairment from tremor in relatives of patients with essential tremor: a community-based family study. Ann Neurol 2001; 49: 761–769.
Louis ED, Dogu O : Does age of onset in essential tremor have a bimodal distribution? Data from a tertiary referral setting and a population-based study. Neuroepidemiology 2007; 29: 208–212.
Gulcher JR, Jonsson P, Kong A et al: Mapping of a familial essential tremor gene, FET1, to chromosome 3q13. Nat Genet 1997; 17: 84–87.
Higgins JJ, Pho LT, Nee LE : A gene (ETM) for essential tremor maps to chromosome 2p22-p25. Mov Disord 1997; 12: 859–864.
Shatunov A, Sambuughin N, Jankovic J et al: Genomewide scans in North American families reveal genetic linkage of essential tremor to a region on chromosome 6p23. Brain 2006; 129: 2318–2331.
Kovach MJ, Ruiz J, Kimonis K et al: Genetic heterogeneity in autosomal dominant essential tremor. Genet Med 2001; 3: 197–199.
Illarioshkin SN, Rakhmonov RA, Ivanova-Smolenskaia IA et al: [Molecular genetic analysis of essential tremor]. Genetika 2002; 38: 1704–1709.
Lucotte G, Lagarde JP, Funalot B, Sokoloff P : Linkage with the Ser9Gly DRD3 polymorphism in essential tremor families. Clin Genet. 2006; 69: 437–440.
Higgins JJ, Loveless JM, Jankovic J, Patel PI : Evidence that a gene for essential tremor maps to chromosome 2p in four families. Mov Disord 1998; 13: 972–977.
Higgins JJ, Lombardi RQ, Pucilowska J, Jankovic J, Tan EK, Rooney JP : A variant in the HS1-BP3 gene is associated with familial essential tremor. Neurology 2005; 64: 417–421.
Merner ND, Girard SL, Catoire H et al: Exome sequencing identifies FUS mutations as a cause of essential tremor. Am J Hum Genet 2012; 91: 313–319.
Parmalee N, Mirzozoda K, Kisselev S et al: Genetic analysis of the FUS/TLS gene in essential tremor. Eur J Neurol 2013; 20: 534–539.
Ortega-Cubero S, Lorenzo-Betancor O, Lorenzo E et al: Fused in sarcoma (FUS) gene mutations are not a frequent cause of essential tremor in Europeans. Neurobiol Aging 2013; 34: e9–2441.e11.
Labbe C, Soto-Ortolaza AI, Rayaprolu S et al: Investigating the role of FUS exonic variants in Essential Tremor. Parkinsonism Relat Disord. 2013; 19: 755–757.
Rajput A, Rajput AH, Rajput ML et al: Identification of FUS p.R377W in essential tremor. Eur J Neurol 2014; 21: 361–363.
Deuschl G, Elbe R : Essential tremor-neurodegenerative or nondegenerative disease towards a working definition of ET. Parkinsonism Mov Disord 2009; 24: 2033–2041.
Louis ED : Essential tremor: a common disorder of Purkinje neurons? Neuroscientist 2015, ; e-pub ahead of print 4 June 2015..
Helmchen C, Hagenow A, Miesner J et al: Eye movement abnormalities in essential tremor may indicate cerebellar dysfunction. Brain 2003; 126: 1319–1332.
Quattrone A, Cerasa A, Messina D et al: Essential head tremor is associated with cerebellar vermisatrophy: a volumetric and voxel-based morphometry MR imaging study. AJNR Am J Neuroradiol 2008; 29: 1692–1697.
Louis ED, Faust PL, Vonsattel JP et al: Neuropathological changes in essential tremor: 33 cases compared with 21 controls. Brain 2007; 130 (Pt 12): 3297–3307.
Louis ED : From neurons to neuron neighborhoods: the rewiring of the cerebellar cortex in essential tremor. Cerebellum 2014; 13: 501–512.
Boecker H : Imaging the role of GABA in movement disorders. Curr Neurol Neurosci Rep. 2013; 13: 385.
Gironell A : The GABA hypothesis in essential tremor: lights and shadows. Tremor Other Hyperkinet Mov (N Y) 2014; 4: 254.
Louis ED, Pullman SL : Comparison of clinical vs. electrophysiological methods of diagnosing of essential tremor. Mov Disord 2001; 16: 668–673.
Louis ED, Barnes L, Wendt KJ et al: A teaching videotape for the assessment of essential tremor. Mov Disord 2001; 16: 89–93.
Louis ED, Ford B : Bismuth B. Reliability between two observers using a protocol for diagnosing essential tremor. Mov Disord 1998; 13: 287–293.
Louis ED, Wendt KJ, Albert SM, Pullman SL, Yu Q, Andrews H : Validity of a performance-based test of function in essential tremor. Arch Neurol 1999; 56: 841–846.
Louis ED, Levy G, Cote LJ, Mejia H, Fahn S, Marder K : Diagnosing Parkinson's disease using videotaped neurological examinations: validity and factors that contribute to incorrect diagnoses. Mov Disord 2002; 17: 513–517.
Louis ED, Ottman R, Ford B et al: The Washington heights-inwood genetic study of essential tremor: methodologic issues in essential-tremor research. Neuroepidemiology 1997; 16: 124–133.
Deuschl G, Bain P, Brin M : Consensus statement of the movement disorder society on tremor. Ad Hoc Scientific Committee. Mov Disord 1998; 13: 2–23.
Li H, Durbin R : Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25: 1754–1760.
McKenna A, Hanna M, Banks E et al: The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010; 20: 1297–1303.
Hu H, Roach JC, Coon H et al: A unified test of linkage analysis and rare-variant association for analysis of pedigree sequence data. Nat Biotechnol 2014; 32: 663–669.
Choi Y, Sims GE, Murphy S, Miller JR, Chan AP : Predicting the functional effect of amino acid substitutions and indels. PloS One 2012; 7: e46688.
Kumar P, Henikoff S, Ng PC : Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009; 4: 1073–1081.
Schwarz JM, Rodelsperger C, Schuelke M, Seelow D : MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 2010; 7: 575–576.
de la Monte SM, Sohn YK, Etienne D, Kraft J, Wands JR : Role of aberrant nitric oxide synthase-3 expression in cerebrovascular degeneration and vascular-mediated injury in Alzheimer's disease. Ann N Y Acad Sci 2000; 903: 61–71.
Abbott LC, Nahm SS : Neuronal nitric oxide synthase expression in cerebellar mutant mice. Cerebellum 2004; 3: 141–151.
de la Monte SM, Lu BX, Sohn YK et al: Aberrant expression of nitric oxide synthase III in Alzheimer's disease: relevance to cerebral vasculopathy and neurodegeneration. Neurobiol Aging 2000; 21: 309–319.
Salinas M, Duprat F, Heurteaux C, Hugnot JP, Lazdunski M : New modulatory alpha subunits for mammalian Shab K+ channels. J Biol Chem 1997; 272: 24371–24379.
Ueda A, Wu CF : Distinct frequency-dependent regulation of nerve terminal excitability and synaptic transmission by IA and IK potassium channels revealed by Drosophila Shaker and Shab mutations. J Neurosci 2006; 26: 6238–6248.
Kaplan WD, Trout WE 3rd : The behavior of four neurological mutants of Drosophila. Genetics 1969; 61: 399–409.
Engel JE, Wu CF : Interactions of membrane excitability mutations affecting potassium and sodium currents in the flight and giant fiber escape systems of Drosophila. J Comp Physiol A 1992; 171: 93–104.
Jan YN, Jan LY, Dennis MJ : Two mutations of synaptic transmission in Drosophila. Proc R Soc Lond B Biol Sci 1977; 198: 87–108.
Klein A, Boltshauser E, Jen J, Baloh RW : Episodic ataxia type 1 with distal weakness: a novel manifestation of a potassium channelopathy. Neuropediatrics 2004; 35: 147–149.
Matsukawa H, Wolf AM, Matsushita S, Joho RH, Knopfel T : Motor dysfunction and altered synaptic transmission at the parallel fiber-Purkinje cell synapse in mice lacking potassium channels Kv3.1 and Kv3.3. J Neurosci 2003; 23: 7677–7684.
McMahon A, Fowler SC, Perney TM, Akemann W, Knöpfel T, Joho RH : Allele-dependent changes of olivocerebellar circuit properties in the absence of the voltage-gated potassium channels Kv3.1 and Kv3.3. Eur J Neurosci 2004; 19: 3317–3327.
Erickson-Davis CR, Faust PL, Vonsattel JP, Gupta S, Honig LS, Louis ED : ‘Hairy baskets’ associated with degenerative Purkinje cell changes in essential tremor. J Neuropathol Exp Neurol 2010; 69: 262–271.
Kuo SH, Tang G, Louis ED et al: Lingo-1 expression is increased in essential tremor cerebellum and is present in the basket cell pinceau. Acta Neuropathol 2013; 125: 879–889.
Bekku Y, Su WD, Hirakawa S et al: Molecular cloning of Bral2, a novel brain-specific link protein, and immunohistochemical colocalization with brevican in perineuronal nets. Mol Cell Neurosci 2003; 24: 148–159.
Bekku Y, Saito M, Moser M et al: Bral2 is indespensible for the proper localization of Brevican and the structural integrity of the perineuronal net in the brainstem and cerebellum. J Comp Neurol 2012; 520: 1721–1736.
Li X, Stevens PD, Yang H et al: The deubiquitination enzyme USP46 functions as a tumor suppressor by controlling PHLPP-dependent attenuation of Akt signaling in colon cancer. Oncogene 2013; 32: 471–478.
Zhang W, Tian QB, Li QK et al: Lysine 92 amino acid residue of USP46, a gene associated with 'behavioral despair' in mice, influences the deubiquitinating enzyme activity. PloS One 2011; 6: e26297.
Wu YR, Chen CM, Chen YC et al: Ubiquitin specific proteases USP24 and USP40 and ubiquitin thiolesterase UCHL1 polymorphisms have synergic effect on the risk of Parkinson's disease among Taiwanese. Clin Chim Acta 2010; 411: 955–958.
Kushima I, Aleksic B, Ito Y et al: Association study of ubiquitin-specific peptidase 46 (USP46) with bipolar disorder and schizophrenia in a Japanese population. J Hum Genet 2010; 55: 133–136.
Tomida S, Mamiya T, Sakamaki H et al: Usp46 is a quantitative trait gene regulating mouse immobile behavior in the tail suspension and forced swimming tests. Nat Genet 2009; 41: 688–695.
Imai S, Mamiya T, Tsukada A et al: Ubiquitin-specific peptidase 46 (Usp46) regulates mouse immobile behavior in the tail suspension test through the GABAergic system. PloS One 2012; 7: e39084.
Acknowledgements
Dr Louis has received research support from the National Institutes of Health (NIH): NINDS #R01 NS042859 (principal investigator), NINDS #R01 NS39422 (principal investigator), NINDS #R01 NS086736 (principal investigator), NINDS #R01 NS073872 (principal investigator), NINDS #R01 NS085136 (principal investigator) and NINDS #R01 NS088257 (principal investigator). Dr Clark is funded by NIH grants R21NS050487 (PI), R01NS060113 (PI), R01NS0738072 (CoPI), P50AG008702 (CoI), P50 NS038370 (CoI), the Parkinson’s disease foundation (PI) and the Michael J Fox foundation (CoI). Dr Ottman is funded by NINDS #R01 NS078419 (PI), #U01 NS077276 (MPI), #U01 NS077367 (MPI), #NIH R01 NS073872 (CoI), NHGRI #P50 HG007257 (CoI), and NIA #R01 AG041797 (CoI). We thank the patients and families for participating in this study. We like to acknowledge and thank Drs Peter Nagy and Jane Dunning Broadbent for performing the sequencing at the clinical NGS core, laboratory of personalized medicine, at Columbia University.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Liu, X., Hernandez, N., Kisselev, S. et al. Identification of candidate genes for familial early-onset essential tremor. Eur J Hum Genet 24, 1009–1015 (2016). https://doi.org/10.1038/ejhg.2015.228
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2015.228
This article is cited by
-
Association Analysis of 27 Single Nucleotide Polymorphisms in a Chinese Population with Essential Tremor
Journal of Molecular Neuroscience (2023)
-
Gene Expression Analysis of Laser-Captured Purkinje Cells in the Essential Tremor Cerebellum
The Cerebellum (2022)
-
Essential tremor
Nature Reviews Disease Primers (2021)
-
Essential tremor pathology: neurodegeneration and reorganization of neuronal connections
Nature Reviews Neurology (2020)
-
A Drosophila Model of Essential Tremor
Scientific Reports (2018)
