
Abstract
The glutamate-induced excitotoxicity pathway has been reported in several neurodegenerative diseases. Molecules that inhibit the release of glutamate or cause the overactivation of glutamate receptors can minimize neuronal cell death in these diseases. Osmotin, a homolog of mammalian adiponectin, is a plant protein from Nicotiana tabacum that was examined for the first time in the present study to determine its protective effects against glutamate-induced synaptic dysfunction and neurodegeneration in the rat brain at postnatal day 7. The results indicated that glutamate treatment induced excitotoxicity by overactivating glutamate receptors, causing synaptic dysfunction and neuronal apoptosis after 4 h in the cortex and hippocampus of the postnatal brain. In contrast, post-treatment with osmotin significantly reversed glutamate receptor activation, synaptic deficit and neuronal apoptosis by stimulating the JNK/PI3K/Akt intracellular signaling pathway. Moreover, osmotin treatment abrogated glutamate-induced DNA damage and apoptotic cell death and restored the localization and distribution of p53, p-Akt and caspase-3 in the hippocampus of the postnatal brain. Finally, osmotin inhibited glutamate-induced PI3K-dependent ROS production in vitro and reversed the cell viability decrease, cytotoxicity and caspase-3/7 activation induced by glutamate. Taken together, these results suggest that osmotin might be a novel neuroprotective agent in excitotoxic diseases.
Similar content being viewed by others


Main
Glutamate, a major excitatory neurotransmitter in the mammalian central nervous system (CNS), is one of the most important neurotransmitters and plays a key role in neural development, synaptic plasticity, learning and memory under physiological conditions.1 Lucas and Newhouse2 reported the toxic effects of glutamate in the retina of mice, and Olney3 demonstrated the concept of glutamate-induced excitotoxicity in the mouse brain. Excitotoxicity is the consequence of imbalances in glutamatergic neurotransmission that leads to neuronal death. Excitotoxicity-mediated neuronal cell death has been reported in various disorders of the CNS, including ischemia and neurodegenerative diseases.4
Previous studies performed both in vitro and in vivo have demonstrated that the overstimulation of NMDA- or AMPA-type glutamate receptors induced neuronal apoptosis.5, 6 An excess of glutamate and glutamatergic activity has been demonstrated in certain neurodegenerative diseases. Choi7 reported calcium-mediated glutamate excitotoxicity in neuronal cultures. The prolonged activation of NMDA receptors triggers an increase in the intracellular calcium load, activates catabolic enzymes and caspases and produces reactive oxygen and nitrogen-free radicals, eventually leading to apoptosis.8, 9 The exact mechanism of glutamate-induced excitotoxicity is not clear, and some believe that this process may be mediated through the activation of mitogen-activated protein kinases (MAPKs) and inhibition of the PI3K/Akt pathway.10, 11, 12, 13 The MAPK family comprises serine/threonine kinases including ERK1/2, c-Jun N-terminal kinases (JNK1/2/3) and p38 MAPK. These kinases are activated downstream of many extracellular signals, including signals from growth factors, cytokines and neurotransmitters, and have key roles in the development of neurons, synaptic plasticity, cell survival and cell death.14, 15, 16, 17 Indeed, several studies have reported that growth factors and neurotransmitters modulate a pathway involving phosphatidylinositol 3-kinase (PI3K) and Akt (otherwise known as PKB) that is of vital importance in cell growth, proliferation, metabolism, neuroplasticity and cell survival.18, 19
Osmotin is a 24-kDa multifunctional plant protein from tobacco (Nicotiana tabacum) and a member of the pathogenesis related-5 (PR-5) family of proteins that provide osmotolerance to plants and exhibit antifungal activity.20, 21 As the structural and functional homolog of the mammalian hormone adiponectin, osmotin is believed to have biological applications as an adiponectin agonist and was shown to reduce the anti-inflammatory effect of adiponectin in a murine colitis model.22, 23, 24 Previous studies have shown that adiponectin was able to protect hippocampal neurons against excitotoxicity induced by kainic acid.25, 26
These findings led us to the present work, and the aims of our study were to investigate the neuroprotective effect of osmotin against glutamate-induced excitotoxicity and neurodegeneration and to determine the mechanism involved in this effect in the cortex and hippocampus of the postnatal day 7 (P7) rat brain.
Results
Osmotin attenuated the glutamate-induced excitotoxicity in the cortex and hippocampus of the developing rat brain
AMPARs are ligand-gated ion channels that consist of combinations of four different subunits: GluR1–4. Glutamate receptor overexpression can lead to excitotoxicity and ultimately neuronal degeneration. A single subcutaneous injection of glutamate significantly induced the overexpression of glutamate receptor (AMPAR, Glu2/3/4) protein levels 4 h after the injection in the cortex and hippocampus of P7 rat brains. A significant decrease was observed in the protein levels of AMPARs in the group of animals treated with osmotin following glutamate treatment (Figure 1a). We also examined the phosphorylation of AMPARs (Glu1) at serine845, as this modification has been reported to be involved in excitotoxic insult and long-term potentiation (LTP). A marked increase in p-AMPAR was observed in glutamate-treated animals in both areas of the developing brain, whereas, as expected, osmotin reversed the glutamate-induced changes in the p-AMPARs in the cortex and hippocampus of the developing brain.

The effect of osmotin on glutamate-induced expression of AMPARs, p-AMPARs, CaMKII, synaptophysin and p-CREB protein. Representative western blots and densitometry histograms depicting (a) AMPARs, p-AMPARs, CaMKII, (b) synaptophysin and p-CREB expression in the cortex and hippocampus of 7-day-old rats after glutamate and osmotin treatment are shown. Sigma Gel software was used for quantifying the protein bands. β-Actin was used as a loading control. All the related experimental details are provided in the Materials and Methods section. The density values are expressed in arbitrary units (AUs) as the mean±S.E.M. for the indicated proteins (n=5 rats/group). *Significantly different from the control; #significantly different from glutamate. Significance=P<0.05
Mitochondrial Ca2+ loading is a critical step in acute glutamate excitotoxicity. Excitotoxicity is associated with marked increases in free intracellular calcium levels.2 Calmodulin-dependent protein kinase-II (CaMKII) is an important regulator of biochemical events that lead to neuronal cell death because of acute excitotoxicity, and inhibition of this kinase by an antagonist has been shown to reduce excitotoxic neuronal cell death.27 The expression level of CaMKII after 4 h of glutamate treatment in the developing brains was examined using western blotting. A marked increase was observed in the expression of the CaMKII protein in both regions of the developing brain. Osmotin treatment after glutamate treatment significantly reversed this elevated level of CaMKII in these regions of the developing brain, suggesting that osmotin has a neuroprotective effect against glutamate-induced acute excitotoxicity (Figure 1a).
Osmotin reversed glutamate-induced synaptic dysfunction in the cortex and hippocampus of the developing rat brain
Several studies have correlated the plasticities of synaptic morphological structures and convective function in neurons at the synapse with changes in proteins, neurotransmitters, receptors and secondary messengers.28, 29 Elevations in glutamate-induced CaMKII and Ca+2 concentrations have adverse effects on neuronal synapses. To analyze the effect of elevated glutamate and the possible reversal effect of osmotin on synaptic plasticity, the expression of synaptophysin in both regions of the developing brain was measured by western blot. Glutamate significantly decreased the number of neuronal synapses (shown in Figure 1b), as demonstrated by the decrease in synaptophysin protein expression, that is widely accepted as a marker for neuronal synapses. However, pups that received osmotin post treatment showed a significantly increased level of synaptophysin compared with the glutamate-treated group, revealing that osmotin reversed the adverse effects of glutamate-induced synaptic plasticity in the cortex and hippocampus of the developing brain.
Next, western blot analysis was used to measure the effect of glutamate treatment on phospho-CREB in the cortex and hippocampus of the developing brain. Compared with the saline-treated group, glutamate-treated animals showed a significant decrease in the levels of p-CREB protein in the cortex and hippocampus of the developing brain (Figure 1b). A significant increase in the expression of the p-CREB protein was observed following the treatment with osmotin after glutamate treatment, suggesting that osmotin might be involved in minimizing LTP in the cortex and hippocampus of the developing brain.
Osmotin attenuated the glutamate-induced neurodegeneration and DNA fragmentation in the cortex and hippocampus of the developing rat brain
A previous study reported that glutamate excitotoxicity insult can induce widespread p53-mediated neurodegeneration.30 For this reason, the levels of different apoptotic markers were examined via western blotting to determine whether 4 h of glutamate treatment (10 mg/kg) induced neuronal degeneration in the cortex and hippocampus of the developing brain. The data in Figure 2a indicate that glutamate induced toxicity by increasing the expression level of proapoptotic p53, the ratio of proapoptotic Bax to antiapoptotic Bcl-2, the translocation of mitochondrial cytochrome c into the cytosol and the activation of caspase-3 and PARP-1 (Figure 2b) in both the cortex and hippocampus of the developing brain. In contrast, osmotin treatment significantly reversed the changes induced by glutamate (i.e., decreased the protein expression of p53 and the ratio of Bax/Bcl-2 in the cortex and hippocampus of the developing brain). Osmotin also reduced the translocation of mitochondrial cytochrome c and the levels of activated caspase-3 and PARP-1 in the developing brain.

The effect of osmotin on glutamate-induced neurodegeneration in the cortex and hippocampus of the developing brain. Representative western blots and densitometry histograms showing (a) p53, the Bax/Bcl-2 ratio, (b) cytochrome c, caspase-3 and PARP-1 in the cortex and hippocampus of 7-day-old rats after glutamate and osmotin treatment are shown. Sigma Gel software was used for the quantification of the protein bands. β-Actin was used as a loading control. The density values are expressed in arbitrary units (AUs) as the mean±S.E.M. for the indicated proteins (n=5 rats/group). *Significantly different from the control; #significantly different from glutamate. Significance=P<0.05
Glutamate-induced apoptosis involves the cleavage of PARP-1. DNA damage is one of the hallmarks of apoptosis. Thus, we used TUNEL (terminal deoxynucleotidyl transferase dUTP nick-end labeling) staining (an assay that identifies DNA breaks based on the enzymatic labeling of free 3′ DNA ends) to visualize damaged DNA. Compared with the control-treated pups, glutamate significantly induced nuclear fragmentation and apoptotic bodies in three regions of the hippocampus, CA1, CA3 and DG, as shown in Figure 3. The treatment with osmotin following glutamate treatment significantly reduced nuclear fragmentation and apoptotic bodies (fewer TUNEL-positive cells) in these three hippocampal regions of the developing brain (Figure 3).

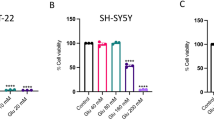
The effect of osmotin on glutamate-induced DNA fragmentation in the hippocampus of the developing brain. (A) The effect of treatment with osmotin on glutamate-induced apoptotic neurodegeneration. Representative photomicrographs of TUNEL staining are shown that reveal apoptotic neuronal cells after glutamate and osmotin treatment. TUNEL-stained apoptotic neurons are shown in the CA1, CA3 and DG regions of the hippocampus. The change in TUNEL-positive cells indicates that osmotin effectively reduced glutamate-induced apoptosis in these three regions of the hippocampus. Images are representative of the staining observed in each section (n=5 animals/group). TUNEL-stained brain tissue at a low magnification ( × 10) and higher magnification ( × 400). TUNEL-positive cells in the different regions of each section were counted using a computer-based program. Significant differences were determined using a one-way analysis of variance (ANOVA) followed by Student’s t-test. *Significantly different from the control (CA1, CA3, DG); #significantly different from glutamate (CA1, CA3, DG); significance=P<0.05. (B) Histogram showing (a) cell viability, (b) cytotoxicity and (c) caspase-3/7 assays performed using the ApoTox-Glo Triplex Assay (Promega, Promega BioSciences, LLC., San Luis Obispo, CA, USA). All the related experimental details are provided in the Materials and Methods section. *Significantly different from the control; #significantly different from glutamate; significance=P<0.05
Effect of osmotin on glutamate-induced neurotoxicity in vitro
To further evaluate the neuroprotective role of osmotin against glutamate in vitro, we measured neuronal HT22 cell viability/cytotoxicity and apoptosis (using the apoptotic marker caspase-3/7) with an Apo-Tox Glo assay. Glutamate treatment resulted in a significant reduction in the number of viable HT22 cells, whereas the cell toxicity and activation of caspase-3/7 significantly increased compared with the control. In contrast, the treatment with osmotin significantly reduced the effects of glutamate, thereby increasing cell viability and decreasing cytotoxicity and caspase-3/7 activation, indicating that osmotin reduced glutamate-induced neurotoxicity (Figure 3Ba–c).
Osmotin protected the developing brain from glutamate-induced neurodegeneration via the JNK/PI3K/Akt intracellular signaling pathways
To further explore the signaling pathways involved in the effects of glutamate and osmotin, western blot analysis was performed to measure phospho-JNK (c-Jun N-terminal kinases) in both glutamate-treated animals and animals that received osmotin after glutamate treatment. Glutamate induced a significant increase in the p-JNK levels in the cortex and hippocampus that was reversed in animals treated with osmotin after glutamate treatment (Figure 4a). The phospho-JNK expression in the hippocampus of developing brains was also examined using immunofluorescence. Interestingly, these immunofluorescence results were consistent with our western blot results. Glutamate increased the expression of p-JNK in the CA1 region of the hippocampus in the developing brain, whereas osmotin treatment decreased p-JNK expression in this region (Figure 4b).

The effect of osmotin on glutamate-induced intracellular JNK/PI3K/Akt signaling in the cortex and hippocampus of the developing brain. (a) Representative western blots and densitometry histograms of the p-JNK/p-PI3K/p-Akt proteins in the cortex and hippocampus of 7-day-old rats after glutamate and osmotin treatment are shown. Sigma Gel software was used for the quantification of the protein bands. β-Actin was used as a loading control. The density values are expressed in arbitrary units (AUs) as the mean±S.E.M. for the indicated proteins (n=5 rats/group). (b) Localization of phospho-JNK and phospho-PI3K in the hippocampus (CA1) of 7-day-old rats after glutamate and osmotin treatment. Images are representative of the staining observed in each section (n=5 animals/group). The immunofluorescence images indicate the localization of p-JNK (green) and p-PI3K (red). Magnification: × 400, Scale bar: 50 μm. (c) Representative western blots and densitometry histograms of p-PI3K, p-Akt, active caspase-3 and PARP-1 proteins in the hippocampus of 7-day-old rats after treatment with glutamate, osmotin and PI3K inhibitor are shown. (d) Histogram illustrating ROS determination in HT22 cells treated with glutamate, osmotin and PI3K inhibitor in vitro. All the related experimental details are provided in the Materials and Methods section. *Significantly different from control; #significantly different from glutamate. Significance=P<0.05
Cantrell31 indicated in his report that the PI3-K/Akt pathway is one of the main prosurvival pathways in neurons. Accordingly, we examined the possible involvement of this pathway following 4 h of glutamate treatment in the cortex and hippocampus of the developing brain. As expected, glutamate significantly decreased the expression of phospho-PI3K and phospho-Akt in both regions of the developing brain as measured by western blot, whereas osmotin significantly reversed this reduction (Figure 4a). Furthermore, the expression of p-PI3K in the hippocampus of the developing brain was examined using immunofluorescence. Confocal microscopy revealed that osmotin reversed the effect of glutamate in the CA1 region of the hippocampus of the developing brain (Figure 4b).
To confirm the involvement of PI3K in this pathway, the PI3K inhibitor LY294002 was administered in vivo to examine its effects on glutamate-induced neurotoxicity. The results showed that LY294002 not only blocked the effects of glutamate on p-PI3K and p-AKT, but also blocked the glutamate-induced expression changes in caspase-3 and PARP-1 (Figure 4c).
The effect of osmotin on glutamate-induced generation of reactive oxygen species (ROS) is PI3K dependent in vitro
To further examine whether the effect of osmotin was mediated via its antioxidant ability, we measured ROS levels in neuronal HT22 cells in vitro. The exposure of cells to glutamate led to a significant increase in ROS levels compared with the control cells, whereas osmotin markedly reduced the ROS level, indicating that osmotin is a potent antioxidant. We then used the PI3K inhibitor LY294002 to confirm that glutamate-induced ROS are PI3K dependent. Our results revealed that glutamate fails to induce ROS after LY294002 treatment, suggesting that the glutamate-induced ROS increase and the osmotin-induced inhibition of ROS are PI3K dependent (Figure 4d).
Osmotin abrogated glutamate-induced distributional changes and colocalization of p53, p-Akt and caspase-3 in the hippocampus of the developing brain
To confirm our western blot results and the proposed mechanism of osmotin-induced neuroprotective effects against glutamate-induced neurodegeneration via p-Akt and p53, we performed experiments to measure the colocalization of p-Akt and p53 and observed their distributional changes in the hippocampus (CA1) of the developing brain. Our results (Figure 5a) indicated that p-Akt was largely colocalized with p53. Compared with the control, the glutamate treatment inhibited phosphorylated Akt (red) and increased the level of p53 (green) in the hippocampal CA1 region of the developing brain. In contrast, osmotin administration reversed the distributional changes in p-Akt (red) and p53 (green) induced by glutamate in the CA1 region of the hippocampus.

The effect of osmotin on glutamate-induced intracellular distribution and colocalization of p-Akt, p53 and caspase-3 in the hippocampus of the developing brain. Intracellular distribution and colocalization of (a) p-Akt and p53 and (b) p53 and caspase-3 in the hippocampus (CA1) of 7-day-old rats after glutamate and osmotin treatment. Images are representative of the staining observed in each section (n=5 animals/group). The immunofluorescence images indicate the localization of p-Akt, caspase-3 (red) and p53 (green). A confocal image of p-Akt and p53 immunofluorescence, showing colocalization in the same neuron. Magnification: × 400, Scale bar: 50 μm. *Significantly different from control; #significantly different from glutamate. Significance=P<0.05
We also examined the colocalization of p53 and caspase-3 and observed the distributional changes of these proteins in the hippocampus following drug treatment. Compared with the results observed in the saline-treated group, the subcutaneous injection of glutamate into P7 animals resulted in high levels of p53 (green) and caspase-3 (red) protein expression, suggesting that these proteins are mainly colocalized. Osmotin-treated animals displayed reduced expression levels of p53 and caspase-3, and colocalization of these proteins was not observed in the hippocampus of the developing brain (Figure 5b).
Discussion
This study demonstrated that osmotin, a homolog of mammalian adiponectin, protected against glutamate-induced neuronal degeneration both in vivo and in vitro. Osmotin, a plant protein from Nicotiana tabacum, not only reversed glutamate-induced synaptic dysfunction and neurodegeneration in the cortex and hippocampus of the developing brain but also inhibited glutamate-induced ROS production and toxicity in HT22 cells via a mechanism involving the PI3K/Akt pathway.
Glutamate receptors play an important physiological role in synaptic plasticity, growth and differentiation; however, their overstimulation induces excitotoxicity. Consistent with previous studies, we observed that glutamate treatment increased the intracellular calcium level and AMPAR expression that further affected the neuronal synapses in the brain of developing rats. We also reported that osmotin treatment not only reversed the glutamate-induced increases in the intracellular calcium level (CaMKII), AMPAR expression and AMPA phosphorylation (Ser845), but also enhanced the expression of the synaptic marker synaptophysin in P7 rat brains (Figures 1a and b). The phosphorylation of AMPARs at serine residues is very important for determining the levels of these receptors at the synaptic membrane. Previous studies have shown that the phosphorylation of GluR1 at Ser845 and Ser831 contributes to LTP and increases cell surface and synaptic AMPAR levels and excitability.32 The serine phosphorylation of AMPARs is presumed to contribute to the hyperexcitability observed in epilepsy, as confirmed by Rakhade et al.33 following experiments with Ser831 and Ser845 knockout mice. After injecting PTZ into P7 mice, these authors observed an increase in AMPAR phosphorylation at Ser831 and Ser845 sites and a significant increase in the postsynaptic scaffolding protein PSD-95, indicating that the phosphorylation of AMPARs at serine residues is involved in synaptic alterations during neonatal seizures. Shepherd and Huganir34 have shown that excitability and LTP can be reduced by blocking these post-translational changes in AMPARs. Our results showing that osmotin upregulated the expression of glutamate-inhibited phospho-CREB in the brain of developing rats also suggest the neuroprotective role of osmotin against glutamate, as numerous studies have shown that phosphorylated CREB can protect against hypoxic ischemic brain insult and influence LTP in the hippocampus.35, 36, 37
The exact mechanisms of glutamate-induced excitotoxic apoptosis are not completely understood; however, the excessive production of ROS, mitochondrial membrane dysfunction and activation of different kinases and apoptotic markers are assumed to play roles in the process.38 Several studies have revealed that G-protein-coupled receptors and group I and II subtypes of metabotropic glutamate receptors can activate the MAPK or PI3K pathways and play key roles in cell proliferation, cell differentiation and cell survival both in vivo and in vitro.39, 40, 41 In addition, the activation of PI3K via numerous neurotransmitters and growth factors has been shown to be followed by Akt phosphorylation at Thr308 and Ser473.42, 43 Several lines of evidence have shown that Akt promotes cell survival and provides protection against glutamate-induced neurotoxicity via the PI3K/Akt/GSK3β (Ser9) pathway.44 Our results have shown that glutamate was mainly involved in the production of ROS and the stimulation of the PI3K/Akt pathway, resulting in neuronal apoptosis. Similarly, glutamate-induced excitotoxicity has been reported to trigger PI3K/Akt activation, whereas drugs capable of reversing PI3K/Akt activation are neuroprotective.45 The current study also demonstrates that osmotin, a plant defense protein and a homolog of human adiponectin, reduced the production of ROS and rescued the cells and developing brain against glutamate through the PI3K/Akt pathway, which was also confirmed using a specific inhibitor of PI3K in vivo and in vitro. The role of osmotin in the reduction of glutamate-induced ROS may be similar to that of adiponectin, which has antioxidant capabilities, as demonstrated by many recent studies.46, 47, 48
A large body of evidence indicates that glutamate treatment induces different apoptotic proteins that play critical roles in neuronal degeneration. Qin et al.49 reported that glutamate receptor overexpression induced the activation of proapoptotic proteins, such as p53, and triggered neuronal injury and death. Furthermore, p53 upregulates the proapoptotic protein Bax and downregulates the antiapoptotic protein Bcl-2, subsequently increasing the release of cytochrome c from mitochondria and triggering caspase-3 activation.49, 50 Our work demonstrated that osmotin attenuated both the glutamate-induced activation of different apoptotic markers and the fragmentation of DNA in the developing brain. These results are also in agreement with other early studies suggesting a neuroprotective role for adiponectin. Nishimura et al.51 reported that adiponectin-deficient mice had enlarged brain infarctions and increased neurological deficits after ischemia/reperfusion compared with wild-type mice, whereas adiponectin treatment increased the protective effects in wild-type mice. An increased adiponectin level in the plasma has also been reported to protect against cardiac ischemia/reperfusion damage in mice.52, 53
In conclusion, our data suggest (Figure 6) that the Nicotiana tabacum protein osmotin has novel neuroprotective effects against glutamate-induced synaptic dysfunction, excitotoxicity and neurodegeneration in the developing brain and that it also reduces the ROS produced by glutamate in vitro. Thus, osmotin may be beneficial in the treatment of neurodegenerative diseases.

A schematic diagram illustrating the proposed mechanism by which osmotin protects against glutamate-induced synaptic plasticity and neurodegeneration in the brain of postnatal rats. Glutamate-induced neurodegeneration is caused by the overactivation of glutamate receptors, followed by the increased release of intracellular Ca2+ that stimulates the JNK/PI3K/Akt pathway, resulting in widespread neurodegeneration via p53 activation. This activated p53 causes an increase in the Bax/Bcl-2 ratio, leading to the translocation of cytochrome c that then activates caspase-3 and PARP-1 expression, inducing DNA fragmentation and cell apoptosis. However, successively treating with osmotin after glutamate successfully enhances synaptic plasticity and inhibits glutamate receptor activation and neurodegeneration through the JNK/PI3K/Akt pathway
Materials and Methods
Animals and drug treatment
Postnatal P7 Sprague–Dawley rats (18 g average body weight) were divided into six groups (n=5 rats per group) that were treated with 0.9% saline (control (C)), 10 mg/kg glutamate (Glu), 15 μg/g osmotin (Os), 10 mg/kg glutamate plus 15 μg/g osmotin (Glu+Os), 10 mg/kg glutamate plus 10 mM LY294002 (Glu+YL) and 10 mg/kg glutamate plus 10 mM LY294002 plus 15 μg/g osmotin (Glu+YL+Os). LY294002 was administered 15 min before the drug treatments, and the animals received this treatment subcutaneously. For post-treatment, osmotin was given 1 and 2 h after glutamate treatment. The animals were killed after 4 to 12 h of treatment. Great care was taken in handling to minimize the suffering of the animals. All experimental and ethical procedures were followed according to the local animal ethics committee of the Division of Applied Life Sciences of the Department of Biology at Gyeongsang National University, South Korea.
Chemicals
Glutamate, the PI3K inhibitor LY294002 and 2′,7′-dichlorofluorescein diacetate (DCFDA) were purchased from Sigma-Aldrich (Saint Louis, MO, USA) and osmotin was purified from salt-adapted cultured tobacco cells as previously described.54 The endotoxin content of the osmotin was <0.03 EU/mg protein.
ApoTox-Glo triplex assay
The ApoTox-Glo Triplex Assay (Promega) was performed to assess viability, cytotoxicity and caspase-3/7 activation within a single assay well. The assay consists of two parts: in the first part, the activities of two proteases, which are markers of cell viability and cytotoxicity, were measured simultaneously. Mouse hippocampal neuronal HT22 cells (2 × 104 cells) were cultured in 96-well assay plates. Each well contained a final volume of 200 μl of DMEM containing 10% FBS and 1% penicillin/streptomycin. After 48 h of incubation at 37 °C in a humidified 5% CO2 incubator, the cells were treated with glutamate (100 μM) and osmotin (0.2 μM). For the assay, 20 μl of the viability/cytotoxicity reagent containing both GF-AFC substrate and bis-AAF-R110 substrate was added to all the wells, briefly mixed via orbital shaking (500 r.p.m. for 30 s) and incubated for 1 h at 37 °C. The fluorescence was measured at two wavelengths: 400 nm/505 nm (viability) and 485 nm/520 nm (cytotoxicity). The GF-AFC substrate enters live cells and is cleaved by a live-cell protease to release AFC. The bis-AAF-R110 substrate does not enter live cells but rather is cleaved by a dead-cell protease to release R110. The live-cell protease activity is restricted to intact viable cells and is measured using a fluorogenic, cell-permeant peptide substrate (glycyl-phenylalanyl-aminofluorocoumarin (GFAFC)). A second fluorogenic, cell-impermeant peptide substrate (bis-alanylalanylphenylalanyl-rhodamine 110 (bis-AAF-R110)) was used to measure the activity of dead-cell proteases that are released from cells that have lost membrane integrity.
The second part of the assay uses a luminogenic caspase-3 substrate, containing the tetrapeptide sequence DEVD, to measure caspase activity, luciferase activity and cell lysis. The caspase-Glo3/7 reagent was added (100 μl) to all the wells and briefly mixed using orbital shaking (500 r.p.m. for 30 s). After incubation for 30 min at room temperature, the luminescence was measured to determine caspase activation.
Oxidative stress (ROS) detection
Mouse hippocampal neuronal HT22 cells were cultured in 96-well plates. Each well contained a final volume of 200 μl of DMEM medium containing 10% FBS and 1% penicillin/streptomycin. After a 48-h incubation at 37 °C in a humidified 5% CO2 incubator, the cells were treated with fresh media containing glutamate (100 μM), glutamate plus osmotin (100+0.2 μM), glutamate plus LY294002 (100+20 μM) or glutamate plus LY294002 plus osmotin (100+20+0.2 μM) for a total of 20 min. The PI3K inhibitor LY294002 was added to the cells 5 min before the glutamate and osmotin treatments. A 600 μM solution of DCFDA (2′,7′-dichlorofluorescein diacetate) dissolved in DMSO/PBS was added to each well and incubated for 30 min. The plates were then read in ApoTox-Glo (Promega) at 488/530 nm.
Tissue collection and sample preparation
Brain sections from saline-, glutamate- or glutamate-plus-osmotin-treated rat pups (n=5 rats per group) were analyzed after 12 h of drug treatment. Transcardial perfusion was performed with 1X phosphate-buffered saline (PBS) followed by 4% ice-cold paraformaldehyde. The brain tissues were post-fixed overnight in 4% paraformaldehyde and subsequently transferred to 20% sucrose until they sank to the bottom of the tube. The brains were frozen in OCT (Tissue-Tek O.C.T. compound medium, Sakura Finetek USA, Inc., Torrance, CA, USA) and then sectioned into 14 μm sections in the coronal plane with a CM 3050S cryostat (Leica, Wetzlar, Germany). The sections were thaw-mounted on Probe-On positively charged slides (Thermo Fisher Scientific Inc., Waltham, MA, USA).
Western blot analysis
To measure the expression levels of different proteins using western blot analysis, the animals were killed after 4 h of drug treatment. The brains were rapidly removed, and the cortex and hippocampus were dissected and frozen on dry ice. Both regions of the brain were homogenized in 0.2 M PBS including a protease inhibitor cocktail. A Bio-Rad protein assay kit (Bio-Rad Laboratories, Hercules, CA, USA) was used to measure the concentration of proteins in the homogenates. Equal amounts of total protein (30 μg per sample) were electrophoresed through a 10–15% SDS-PAGE gel under reducing conditions and transferred to a polyvinylidene difluoride (PVDF) membrane (Santa Cruz Biotechnology, Santa Cruz, CA, USA).
Prestained protein ladders (GangNam-STAIN, iNtRon Biotechnology, Inc., Kyungki-Do, Republic of Korea) covering a broad range of molecular weights (10–245 kDa) were run in parallel and used to determine the molecular weights of the detected proteins. To block the membrane and minimize nonspecific binding, the membranes were incubated with 5% (w/v) skim milk. Antibodies used in immunoblotting for detecting proteins included rabbit-derived anti-actin, anti-Bcl-2, anti-Bax and anti-caspase-3; goat-derived anti-cytochrome c and anti-p53; and mouse-derived polyclonal p-JNK, and anti-PARP-1 polyclonal antibodies from Santa Cruz Biotechnology. We also used rabbit-derived anti-p-CREB (Ser133), anti-CaMKII, anti-p-Akt (Ser473), anti-p-PI3K (Y458/Y199), anti-p-JNK (Thr183/Tyr185), anti-AMPA, anti-p-AMPA (Ser845) and anti-synaptophysin from Cell Signaling Technology, Inc. (Danvers, MA, USA). Membrane-bound secondary antibodies were visualized using the ECL detection reagent according to the manufacturer’s instructions (Amersham Pharmacia Biotech, Uppsala, Sweden). The X-ray films were scanned, and the optical densities of the bands were analyzed by densitometry using the computer-based Sigma Gel program, version 1.0 (SPSS, Chicago, IL, USA).
Immunofluorescence assays
Tissue-containing slides were washed twice for 15 min in 0.01 M PBS and proteinase K solution was then added to the tissue and incubated for 5 min at 37 °C. Subsequently, the tissues were incubated for 90 min in blocking solution containing normal swine serum and 0.3% Triton X-100 in PBS. Primary antibodies (rabbit polyclonal p-Akt, p-PI3K and caspase-3 from Cell Signaling Technology and goat polyclonal IgG p53 and mouse polyclonal p-JNK from Santa Cruz Biotechnology; all 1 : 100 in PBS) were applied at 4 °C overnight. On the following day, secondary antibodies (swine anti-rabbit TRITC from Dako Denmark A/S (Glostrup, Denmark) and rabbit anti-goat and goat anti-mouse FITC from Santa Cruz Biotechnology; both 1 : 50 in PBS) were applied at room temperature for 90 min. The slides were washed twice with PBS for 5 min. For double staining, the incubations were performed in parallel. Glass coverslips were mounted on glass slides with mounting medium. Images were captured using a confocal microscope (FluoView FV 1000; Olympus, Tokyo, Japan).
TUNEL staining
For the detection of glutamate-induced apoptosis, nuclear DNA was stained with TUNEL (GenScript USA Inc., Piscataway, NJ, USA) and counterstained using PI. TUNEL staining was performed according to the recommendations of the In Situ Cell Death Detection Kit Fluorescein (GenScript). Glass coverslips were mounted onto the glass slides with mounting medium. The FITC filter was used was to detect TUNEL staining (green). TUNEL-positive staining patterns were acquired with a confocal laser scanning microscope (FluoView FV 1000; Olympus). TUNEL-positive cells in the different regions of each section were counted using a computer-based program.
Data analysis and statistics
Bands on the western blots were scanned and analyzed by densitometry using the Sigma Gel System (SPSS Inc.). A computer-based program was used to measure the integrated densities of immunofluorescence images. The density values are expressed as the mean±S.E.M. A one-way analysis of variance (ANOVA) was used to determine significant differences, and Student’s t-test was used to determine significant differences between relevant treatment groups. The P-values of <0.05 were considered significant.
Abbreviations
- CNS:
-
central nervous system
- MAPK:
-
mitogen-activated protein kinase
- JNK:
-
c-Jun N-terminal kinase
- PI3K:
-
phosphatidylinositol 3-kinase
- PR-5:
-
pathogenesis related-5
- LTP:
-
long-term potentiation
- CaMKII:
-
calmodulin-dependent protein kinase-II
- TUNEL:
-
terminal deoxynucleotidyl transferase dUTP nick-end labeling
- ROS:
-
reactive oxygen species
- PBS:
-
phosphate-buffered saline
- PVDF:
-
polyvinylidene difluoride
References
Suzuki M, Nelson AD, Eickstaedt JB, Wallace K, Wright LS, Svendsen CN . Glutamate enhances proliferation and neurogenesis in human neural progenitor cell cultures derived from the fetal cortex. Eur J Neurosci 2006; 24: 645–653.
Lucas DR, Newhouse JP . The toxic effect of sodium L-glutamate on the inner layers of the retina. AMA Arch Ophthalmol 1957; 58: 193–201.
Olney JW . Brain lesions, obesity, and other disturbances in mice treated with monosodium glutamate. Science 1969; 164: 719–721.
Bruijn LI, Miller TM, Cleveland DW . Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci 2004; 27: 723–749.
Shehadeh J, Fernandes HB, Zeron Mullins MM, Graham RK, Leavitt BR, Hayden MR et al. Striatal neuronal apoptosis is preferentially enhanced by NMDA receptor activation in YAC transgenic mouse model of Huntington disease. Neurobiol Dis 2006; 21: 392–403.
Zeron MM, Fernandes HB, Krebs C, Shehadeh J, Wellington CL, Leavitt BR et al. Potentiation of NMDA receptor-mediated excitotoxicity Linked with intrinsic apoptotic pathway in YAC transgenic mouse model of Huntington’s disease. Mol Cell Neurosci 2004; 25: 469–479.
Choi DW . Glutamate neurotoxicity in cortical cell culture is calcium dependent. Neurosci Lett 1985; 58: 293–297.
Jung KH, Chu K, Lee ST, Park HK, Kim JH, Kang KM et al. Augmentation of nitrite therapy in cerebral ischemia by NMDA receptor inhibition. Biochem Biophys Res Commun 2009; 378: 507–512.
Fan MMY, Raymond LA . N-Methyl-D-aspartate (NMDA) receptor function and excitotoxicity in Huntington’s disease. Neurobiology 2007; 81: 272–293.
Yang DD, Kuan CY, Whitmarsh AJ, Rincon M, Zheng TS, Davis RJ et al. Absence of excitotoxicity induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature 1997; 389: 865–870.
Molz S, Decker H, Dal-Cim T, Cremonez C, Cordova FM, Leal RB et al. Glutamate induced toxicity in hippocampal slices involves apoptotic features and p38MAPK signaling. Neurochem Res 2008b; 33: 27–36.
Chuang DM, Wang Z, Chiu CT . GSK-3 as a target for lithium-induced neuroprotection against excitotoxicity in neuronal cultures and animal models of ischemic stroke. Front Mol Neurosci 2011; 4: 15.
Molz S, Dal-Cim T, Budni J, Martín-de-Saavedra MD, Egea J, Romero A et al. Neuroprotective effect of guanosine against glutamate-induced cell death in rat hippocampal slices is mediated by the phosphatidylinositol-3 kinase/Akt/glycogen synthase kinase 3b pathway activation and inducible nitric oxide synthase inhibition. J Neurosci Res 2011; 89: 1400–1408.
Thomas GM, Huganir RL . MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci 2004; 5: 173–183.
Sweatt JD . Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol 2004; 14: 1–7.
Waetzig V, Herdegen T . Neurodegenerative and physiological actions of c- Jun N-terminal kinases in the mammalian brain. Neurosci Lett 2004; 361: 64–67.
Kim EK, Choi EJ . Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta 2010; 1802: 396–405.
Hanada M, Feng J, Hemmings BA . Structure, regulation and function of PKB/AKT – a major therapeutic target. Biochim Biophys Acta 2004; 1697: 3–16.
Brazil DP, Yang ZZ, Hemmings BA . Advances in protein kinase B signalling: AKTion on multiple fronts. Trends Biochem Sci 2004; 29: 23–42.
Cornelissen BJC, Hooft van Huijsduijnen RA, Bol JFA . Tobacco mosaic virus-induced tobacco protein is homologous to the sweet-tasting protein thaumatin. Nature 1986; 321: 531–532.
Abad LR, D’Urzo MP, Liu D, Narasimhan ML, Reuveni M, Zhu JK et al. Antifungal activity of tobacco osmotin has specificity and involves plasma membrane permeabilization. Plant Sci 1996; 118: 11–23.
Narashimhan ML, Coca MA, Jin J, Yamauchi T, Ito Y, Kadowaki T et al. Osmotin is a homolog of mammalian adiponectin and controls apoptosis in yeast through a homolog of mammalian adiponectin receptor. Mol Cell 2005; 17: 171–180.
Miele M, Costantini S, Colonna G . Structural and functional similarities between osmotin from Nicotiana Tabacum seeds and human adiponectin. PLoS One 2011; 6: 16690.
Arsenescu V, Narasimhan ML, Halide T, Bressan RA, Barisione C, Cohen DA et al. Adiponectin and plant-derived mammalian adiponectin homolog exert a protective effect in murine colitis. Dig Dis Sci 2011; 56: 2818–2832.
Qui G . Adiponectin protects in rat hippocampal neurons against excitotoxity. Age 2011; 33: 155–165.
Jeon BT, Shin HJ, Kim JB, Kim YK, Lee DH, Kim KH et al. Adiponectin protects hippocampal neurons against kainic acid-induced excitotoxicity. Brain Res Rev 2009; 61: 81–88.
Takano H, Sugimura M, Kanazawa Y, Uchida T, Morishima Y, Shirasaki Y . Protective effect of DY-9760e, a calmodulin antagonist, against neuronal cell death. Biol Pharm Bull 2004; 27: 1788–1791.
O’Rourke NA, Weiler NC, Micheva KD, Smith SJ . Deep molecular diversity of mammalian synapses: why it matters and how to measure it. Nat Rev Neurosci 2012; 13: 365–379.
Sheng M, Kim MJ . Postsynaptic signaling and plasticity mechanisms. Science 2002; 298: 776–780.
Qin ZH, Tao LY, Chen X . Dual roles of NF-κB in cell survival and implications of NF-κB inhibitors in neuroprotective therapy. Acta Pharmacol Sin 2007; 28: 1859–1872.
Cantrell DA . Phosphoinositide 3-kinase signaling pathways. J Cell Sci 2011; 114: 1439–1445.
He K, Goel A, Ciarkowski CE, Song L, Lee HK . Brain area specific regulation of synaptic AMPA receptors by phosphorylation. Commun Integr Biol 2011; 4: 569–572.
Rakhade SN, Fitzgerald EF, Klein PM, Zhou C, Sun H, Huganir RL et al. Glutamate receptor 1 phosphorylation at serine 831 and 845 modulates seizure susceptibility and hippocampal hyperexcitability after early life seizures. J Neurosci 2012; 32: 17800–17812.
Shepherd JD, Huganir RL . The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol 2007; 23: 613–643.
Impey S, Obrietan K, Storm DR . Making new connections: role of ERK/MAP kinase signaling in neuronal plasticity. Neuron 1999; 23: 11–14.
Bourtchuladze RB, Frenguelli J, Blendy D, Cioffi G, Schutz A, Silva J . Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell 1994; 79: 59–68.
Matthies H, Schulz S, Thiemann W, Siemer H, Schmidt H, Krug M et al. Design of a multiple slice interface chamber and application for resolving the temporal pattern of CREB phosphorylation in hippocampal long-term potentiation. J Neurosci Methods 1997; 78: 173–179.
Ginnan R, Pfleiderer PJ, Pumiglia K, Singer HA . PKCdelta and CaMKII-delta 2 mediate ATP-dependent activation of ERK1/2 in vascular smooth muscle. Am J Physiol Cell Physiol 2004; 286: C1281–C1289.
Schinkmann KA, Kim TA, Avraham S . Glutamate-stimulated activation of DNA synthesis via mitogen-activated protein kinase in primary astrocytes: involvement of protein kinase C and related adhesion focal tyrosine kinase. J Neurochem 2000; 74: 1931–1940.
Karim F, Wang CC, Gereau RW . Metabotropic glutamate receptor subtypes 1 and 5 are activators of extracellular signal-regulated kinase signalling required for inflammatory pain in mice. J Neurosci 2001; 21: 3771–3779.
Marinissen MJ, Gutkind JS . G protein-coupled receptors and signalling networks: emerging paradigms. Trends Pharmacol Sci 2001; 22: 368–376.
Choudhari SR, Khan MA, Harris G, Picker D, Jacob GS, Block T et al. Deactivation of Akt and STAT3 signaling promotes apoptosis, inhibits proliferation, and enhances the sensitivity of hepatocellular carcinoma cells to an anticancer agent. Atiprimod Mol Cancer Ther 2007; 6: 112–121.
Jing Y, Liu LZ, Jiang Y, Zhu Y, Guo NL, Barnett J et al. Cadmium increases HIF-1 and VEGF expression through ROS, ERK, and AKT signaling pathways and induces malignant transformation of human bronchial epithelial cells. Toxicol Sci 2012; 125: 10–19.
Kim Y, Seger R, Suresh babu CV, Hwang SY, Yoo YS . A positive role of the PI3K/Akt signaling pathway in PC12 cell differentiation. Mol Cells 2004; 18: 353–359.
Dal-Cim T, Martins WC, Santos AR, Tasca CI . Guanosine is neuroprotective against oxygen/glucose deprivation in hippocampal slices via large conductance Ca2+-activated K+ channels, phosphatidilinositol-3 kinase/ protein kinase B pathway activation and glutamate uptake. Neuroscience 2011; 183: 212–220.
Essick EE, Wilson RM, Pimentel DR, Shimano M, Baid S, Ouchi N et al. Adiponectin modulates oxidative stress-induced autophagy in cardiomyocytes. PLoS One 2013; 8: e68697.
Fujii Y, Okada A, Yasui T, Niimi K, Hamamoto S, Hirose M et al. Effect of adiponectin on kidney crystal formation in metabolic syndrome model mice via inhibition of inflammation and apoptosis. PLoS One 2013; 8: e61343.
Chan KH, Lam KSL, Cheng OY, Kwan JSC, Ho PWL, Cheng KKY et al. Adiponectin is protective against oxidative stress induced cytotoxicity in amyloid-beta neurotoxicity. PLoS One 2012; 7: e52354.
Qin ZH, Chen RW, Wang Y, Nakai M, Chuang DM, Chase TN . NF-κB nuclear translocation up-regulates c-Myc and p53 during N-methyl-D-aspartate receptor-mediated apoptosis. J Neurosci 1999; 19: 4023–4033.
Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M et al. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004; 303: 1010–1014.
Nishimura M, Izumiya Y, Higuchi A, Shibata R, Qiu J, Kudo C et al. Adiponectin prevents cerebral ischemic injury through endothelial nitric oxide synthase dependent mechanisms. Circulation 2008; 117: 216–223.
Gonon AT, Widegren U, Bulhak A, Salehzadeh F, Persson J, Sjoquist PO et al. Adiponectin protects against myocardial ischaemia-reperfusion injury via AMP-activated protein kinase, Akt, and nitric oxide. Cardiovasc Res 2008; 78: 116–122.
Shinmura K, Tamaki K, Saito K, Nakano Y, Tobe T, Bolli R . Cardioprotective effects of short-term caloric restriction are mediated by adiponectin via activation of AMP-activated protein kinase. Circulation 2007; 116: 2809–2817.
Singh N K, Bracker CA, Hasegawa PM, Handa AK, Buckel S, Hermodson MA et al. Characterization of osmotin. Plant Physiol 1987; 85: 529–536.
Acknowledgements
This research was supported by the Pioneer Research Center Program through the National Research Foundation of Korea, funded by the Ministry of Science, ICT and Future Planning (2012-0009521) and NRF funded by the Korean Government (MSIP; 2013R1A2A1A01005170).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by A Verkhratsky
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Shah, S., Lee, H., Bressan, R. et al. Novel osmotin attenuates glutamate-induced synaptic dysfunction and neurodegeneration via the JNK/PI3K/Akt pathway in postnatal rat brain. Cell Death Dis 5, e1026 (2014). https://doi.org/10.1038/cddis.2013.538
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2013.538
Keywords
This article is cited by
-
Exploring the chemical components of Kuanchang-Shu granule and its protective effects of postoperative ileus in rats by regulating AKT/HSP90AA1/eNOS pathway
Chinese Medicine (2024)
-
GDF1 ameliorates cognitive impairment induced by hearing loss
Nature Aging (2024)
-
Neuroprotective effects of osmotin in Parkinson’s disease-associated pathology via the AdipoR1/MAPK/AMPK/mTOR signaling pathways
Journal of Biomedical Science (2023)
-
Cerebrolysin reduces excitotoxicity by modulation of cell-death proteins in delayed hours of ischemic reperfusion injury
Metabolic Brain Disease (2023)
-
Zinc Ortho Methyl Carbonodithioate Improved Pre and Post-Synapse Memory Impairment via SIRT1/p-JNK Pathway against Scopolamine in Adult Mice
Journal of Neuroimmune Pharmacology (2023)
