
Abstract
Normal tissue damage limits the efficacy of anticancer therapy. For anthracyclines, the clinically most relevant adverse effect is cardiotoxicity. The mechanisms involved are poorly understood and putative cardioprotectants are controversially discussed. Here, we show that the lipid-lowering drug lovastatin protects rat H9c2 cardiomyoblasts from doxorubicin in vitro. Protection by lovastatin is related to inhibition of the Ras-homologous GTPase Rac1. It rests on a reduced formation of DNA double-strand breaks, resulting from the inhibition of topoisomerase II by doxorubicin. Doxorubicin transport and reactive oxygen species are not involved. Protection by lovastatin was confirmed in vivo. In mice, lovastatin mitigated acute doxorubicin-induced heart and liver damage as indicated by reduced mRNA levels of the pro-fibrotic cytokine connective tissue growth factor (CTGF) and pro-inflammatory cytokines, respectively. Lovastatin also protected from doxorubicin-provoked subacute cardiac damage as shown by lowered mRNA levels of CTGF and atrial natriuretic peptide. Increase in the serum concentration of troponin I and cardiac fibrosis following doxorubicin treatment were also reduced by lovastatin. Whereas protecting the heart from harmful doxorubicin effects, lovastatin augmented its anticancer efficacy in a mouse xenograft model with human sarcoma cells. These data show that statins lower the incidence of cardiac tissue injury after anthracycline treatment in a Rac1-dependent manner, without impairing the therapeutic efficacy.
Similar content being viewed by others


Main
Anthracycline derivatives such as doxorubicin rank among the most potent antineoplastic drugs.1 However, an unresolved problem associated with their use is normal tissue damage, in particular their dose-dependent acute and delayed cardiotoxicity, which manifests as congestive heart failure due to cardiomyopathy in up to 20% of the patients.2, 3, 4 The formation of reactive oxygen species (ROS), generation of peroxynitrite by nitric oxide synthases (NOS) and/or inhibition of topoisomerase II (topo II) are discussed to be responsible for the cardiotoxic side effect of doxorubicin.4, 5, 6 Co-administration of anthracyclines with the antioxidant dexrazoxane as a cardioprotectant is in dispute because the molecular mechanisms involved in anthracycline-induced cardiotoxicity, as well as the molecular effects of dexrazoxane are still unclear.7, 8, 9 Moreover, it is under debate whether dexrazoxane has negative effects on the antitumor efficacy of anthracyclines.10, 11 Doxorubicin-induced cardiac dysfunction has been related to the induction of iNOS leading to an increase in highly cytotoxic peroxynitrite.12 Yet, pharmacological targeting of iNOS-mediated NO production as cardioprotective strategy for anthracycline treatments is controversially discussed. This is because mitochondrial damage and cardiotoxicity provoked by doxorubicin were reported to be even enhanced in iNOS knockouts.13, 14 Furthermore, unselective NOS inhibition by N-nitro-L-arginine methyl ester increased doxorubicin-induced mortality in mice.15 Therefore, the development of alternative cardioprotective strategies for anthracycline-based anticancer therapy is of high importance.16
HMG-CoA reductase inhibitors (statins), which are clinically established for lipid-lowering purposes, are known to have pleiotropic biological effects.17 These pleiotropic effects are mainly based on the inhibition of the prenylation of low-molecular weight GTPases that are having key roles in the regulation of numerous cellular processes.17, 18, 19 High doses of statins enhance the tumor cell kill of anticancer drugs20, 21 by impairing G1–S transition22 and triggering apoptosis.23, 24 Furthermore, statins promote TNFα-triggered apoptosis25 and reverse chemoresistance by inhibition of NF-κB.26 Apart from increasing the antitumor activity of numerous anticancer drugs in vitro and in vivo,19 low doses of statins reduce cell death of primary human endothelial cells (HUVECs) provoked by doxorubicin and ionizing radiation in vitro.27, 28 Furthermore, statins protect the intestine from the side effects of radiotherapy in vivo.29, 30, 31 These reports point to a dual beneficial function of statins in anticancer therapy, that is, the promotion of anticancer drug-induced killing of malignant cells and concomitant protection of normal cells. Based on these reports and the well-documented cardioprotective function of statins,32 we hypothesized that statins might be clinically useful for alleviating adverse effects of anthracyclines, in particular anthracycline-induced cardiotoxicity. To scrutinize this hypothesis, we investigated the effect of lovastatin on doxorubicin-induced damage to cardiomyocytes using both in vitro and in vivo model systems. Here, we show that the HMG-CoA reductase inhibitor lovastatin significantly protects against doxorubicin-induced cardiac toxicity and, at the same time, augments the anticancer effect of doxorubicin.
Results
Lovastatin reduces doxorubicin-induced cell death in vitro
To investigate whether statins are able to protect cardiomyoblasts against doxorubicin-induced cytotoxicity, we performed in vitro experiments using the well-established rat H9c2 cardiomyoblast system. Doxorubicin induced a dose-dependent increase in the frequency of apoptotic cells (Figure 1a). Lovastatin clearly protected H9c2 cells from doxorubicin-induced apoptosis (Figure 1a). Measuring cell viability, the 50% inhibitory concentration (IC50) of doxorubicin increased from 1.5 to 5.0 μM in the presence of the statin, confirming the protective effect of lovastatin. To elucidate the molecular mechanisms involved in the anti-apoptotic statin effect, the impact of lovastatin on cellular uptake of doxorubicin (influx) and its removal (efflux) was monitored. Doxorubicin influx and efflux were not affected by lovastatin (Figure 1b). Correspondingly, the mRNA expression levels of the drug transporters mdr-1 and mrp-1 were not enhanced by the statin (Figure 1c). The anti-apoptotic statin effect is likely not related to changes in cell-cycle progression because lovastatin did not impact the number of cells arrested in G2/M phase after doxorubicin exposure and, furthermore, did not influence the level of Chk-1 phosphorylation stimulated by the anthracycline (Supplementary Figure S1).

Lovastatin protects rat cardiomyoblasts from doxorubicin-induced cell death. H9c2 cells were left untreated (−Lova) or were pre-treated overnight with lovastatin (+Lova) (20 μM) before pulse treatment (1 h) with the indicated concentrations of doxorubicin was performed. (a) Incubation was continued for 72 h in the presence of lovastatin (1 μM). The percentage of apoptotic cells was analyzed by FACS-based determination of Annexin-V-positive cells. Results presented are the mean±S.E.M. from four independent experiments. (b) To measure doxorubicin transport, samples were taken for FACS analysis before the pulse treatment (Control), directly after the pulse (+Doxo (1 μM, 1 h)) and 6 h after pulse treatment. The inherent fluorescence of doxorubicin was used to monitor its uptake. The shift to the right from peak 1 (untreated control) to peak 2 reflects the increase in intracellular fluorescence which is due to doxorubicin uptake. To determine doxorubicin efflux, doxorubicin pre-loaded cells were postincubated for 6 h in the absence of the drug. The decrease in cellular fluorescence occurring during this time (that is, the shift from peak 2 to peak 3) represents the efflux of doxorubicin. (c) The mRNA level of the major drug transporters mdr-1 and mrp-1 was analyzed before doxorubicin treatment by quantitative real-time RT-PCR analysis. Results presented are the mean±S.D. from three independent experiments
Lovastatin protects from doxorubicin-induced genotoxicity
Induction of DNA damage, notably DNA double-strand breaks (DSBs), represents the major mechanism by which anthracyclines provoke cell death.33 DNA damage triggers a stress response referred to as the DNA damage response (DDR).34 Phosphorylated H2AX (γH2AX) is a frequently used marker of DNA damage,35 in particular DNA DSBs.36, 37 H2AX phosphorylation was analyzed by western blot analysis and, additionally, by the detection of γH2AX foci by immunohistochemistry. Western blot analysis showed that doxorubicin increased the level of γH2AX. Lovastatin pre-treatment significantly attenuated this response (Figure 2a). Immunohistochemical analysis of γH2AX foci formation showed similar results (Figure 2b). The same holds true for assaying the formation of DSBs by the neutral comet assay (Figure 2c). The number of DNA single-strand breaks, as monitored by the alkaline comet assay, was only marginally affected by the statin (Figure 2d). Based on these data we conclude that lovastatin protects H9c2 cardiomyoblasts from early DNA damage induction (that is, formation of DSBs) by doxorubicin. Anthracycline-induced DSBs are known to mainly result from the inhibition of topoisomerase II (topo II).8, 38 Therefore it is important to note that the expression levels of topo IIα and topo IIβ protein were not altered by the statin (Figure 2e). Furthermore, the protein level of topo II isoforms was not affected by doxorubicin, neither in the presence nor absence of lovastatin (Figure 2f). Hence, the geno-protective effect of lovastatin is independent of changes in topo II protein expression.

Lovastatin protects H9c2 cells from doxorubicin-induced genotoxicity. Untreated (−Lova) or lovastatin (+Lova) (20 μM, overnight) pre-treated cells were exposed to doxorubicin (1 μM). (a) H2AX phosphorylation was analyzed 1 h after doxorubicin treatment by western blot. The relative γH2AX level in untreated control was set to 1.0. Signal intensities were quantified densitrometrically. The histogram shows the mean±S.D. from six independent experiments. *P≤0.05, significant difference as compared with the Doxo-treated cells. (b) The formation of γH2AX foci per nucleus was analyzed by immunohistochemistry 2 h after doxorubicin treatment. Microscopic images displayed are representative. The histogram shows the mean±S.D. from three independent experiments. *P≤0.05, significantly different from Doxo-treated cells. (c) Formation of DSBs was analyzed by the neutral comet assay 2 h after treatment. For control, cells were irradiated with 10 Gy (IR). Shown is the increase in tail DNA as compared with untreated control. The histogram shows the mean±S.D. obtained from five independent experiments. *P≤0.05, significantly different from Doxo-treated cells. (d) The level of DNA strand breaks was analyzed by the alkaline comet assay. For control, cells were irradiated with 5.5 Gy (IR). The histogram shows the mean±S.D. from three independent experiments. ns, difference from Doxo-treated cells is statistically not significant. (e) The expression of Topo IIα and Topo IIβ protein was analyzed by western blot. ERK2 protein expression was determined as internal loading control. Con, untreated control (f) 1 h and 2 h after doxorubicin treatment, the protein level of topoisomerases was analyzed by western blot
Protection by lovastatin is independent of reactive oxygen species
The formation of ROS and/or inhibition of topo II are thought to be responsible for the cardiotoxicity of doxorubicin.4, 5 At the time when H2AX phosphorylation occurred in our experimental setting, ROS formation was not detectable (Figures 3a and b). Furthermore, pre-treatment of H9c2 cardiomyocytes with three different antioxidants (that is, curcumin, resveratrol and N-acetylcysteine) did not significantly constrain γH2AX formation stimulated by doxorubicin (Figure 3c). These data indicate that the geno-protective effect of lovastatin rests on the inhibition of doxorubicin-mediated effects on topo II rather than an interference with the formation or detoxification of ROS. In line with this, H2AX phosphorylation provoked by the specific topo II inhibitor etoposide, which does not produce ROS, was also attenuated by lovastatin (Figure 3d). The protective effect of lovastatin appears to be specific for topo II poisons as it does not apply to ionizing radiation (Figure 3e).

Lovastatin-mediated inhibition of doxorubicin-induced H2AX phosphorylation is independent of ROS. (a and b) H9c2 cells were treated with lovastatin (20 μM; overnight). Afterwards, cells were loaded with DCFDA (1 μM; 30 min) before a doxorubicin pulse (1 μM, 1 h) was applied. Intracellular ROS formation was monitored by microscopy (a) or FACS analysis (b). H2O2 was used as positive control. (c) H9c2 cells were pre-treated with the antioxidants curcumin (Cuc; 12.5 μM), resveratrol (Res; 10 μM) or N-acetylcysteine (NAc; 10 mM) for 1 h. Afterwards, doxorubicin (1 μM) was added and phosphorylation of H2AX was analyzed 1 h later by western blot. The histogram shows the mean±S.D. obtained from the densitometrical quantitation of three independent experiments. Relative γH2AX level in untreated cells (Con) was set to 1.0. The differences observed in the presence of the antioxidants were not significantly different from the doxorubicin only treated cells. (d and e) H9c2 cells were exposed to etoposide (7.5 μM) (d) or were irradiated with 5 Gy (IR) (e) and H2AX phosphorylation was monitored 1 h later by western blot. Quantitative data (mean±S.D.) shown under d result from three independent experiments. *P≤0.05, statistically significant difference from Eto control
Rac1 is involved in the doxorubicin-induced DNA damage response
Protection from doxorubicin-induced DNA damage response was also observed upon inhibition of geranylgeranyltransferase, but not following inhibition of farnesyltransferase (Figure 4a). Furthermore, specific inactivation of the Rho GTPase Rac1 by the clostridial toxins TcsL and TcdBF39, 40 also attenuated doxorubicin-induced H2AX phosphorylation (Figure 4a). In addition, the Rac1 inhibitor NSC23766 also mitigated doxorubicin-triggered phosphorylation of H2AX (Figure 4b). Apparently, Rac1 is essential for this genotoxic stress response provoked by doxorubicin. Rac1-mediated signaling to H2AX partially depends on the activity of stress-activated protein kinases (SAPK/JNK), as pharmacological inhibition of this kinases reduced doxorubicin-induced H2AX phosphorylation (Figure 4c). Inhibition of p38 kinase increased the basal level of phosphorylated H2AX by its own (Figure 4c). Doxorubicin exposure in the presence of p38 kinase inhibitor (p38i) further increased the level of phosphorylated H2AX, although this increase was lower than observed in the absence of p38i (Figure 4c). Collectively, Rac1-regulated and SAKP/JNK-related signaling impact γH2AX phosphorylation following doxorubicin treatment.

Rac1 in involved in doxorubicin-stimulated DDR. (a) H9c2 cells were pre-treated with lovastatin (Lova) (20 μM), geranylgeranyl transferase inhibitor (GGTI) (20 μM) or farnesyl transferase inhibitor (FTI) (2 μM) for 16 h. Pre-treatment with Clostridium difficile toxin BF (TcdBF) (1 μg/ml) or Clostridium sordellii lethal toxin (TcsL) (10 μg/ml) was carried out for 2 h. Afterwards, doxorubicin (1 μM) was added and H2AX phosphorylation (γH2AX) was analyzed 1 h later by western blot. The histogram shows quantitative data (mean±S.D.) which were obtained from three independent experiments. Relative γH2AX level in untreated cells (Con) was set to 1.0. *P≤0.05, significantly different from Doxo-treated cells. (b) H9c2 cells were pre-treated with the Rac1-GEF inhibitor NSC23766 (100 μM, 3 h) before doxorubicin exposure (1 μM). H2AX phosphorylation was assayed 1 h later. (c) After 1 h pre-treatment with p38 kinase inhibitor SB203580 (p38i) (10 μM) or JNK inhibitor (JNKi) (10 μM), doxorubicin (1 μM) was added. After further incubation period of 1 h, phosphorlyation of H2AX was analyzed by western blot. The histogram shows quantitative data (mean±S.D.) obtained from four independent experiments. Relative γH2AX level in untreated cells (Con) was set to 1.0. *P≤0.05, significantly different from Doxo-treated cells
Doxorubicin-induced acute and subacute heart damage are reduced by lovastatin
On the basis of our in vitro findings, we hypothesized that lovastatin may reduce cardiotoxicity, which is the major dose-limiting side effect of doxorubicin in cancer patients. To scrutinize this hypothesis, Balb/c mice, either untreated or pre-treated with lovastatin were exposed to a single dose of doxorubicin and acute heart damage was analyzed 48 h later by measuring the mRNA levels of pro-inflammatory and pro-fibrotic cytokines. We observed that the mRNA expression of the pro-fibrotic connective tissue growth factor (CTGF) was clearly enhanced by doxorubicin. Lovastatin blocked this doxorubicin-stimulated pro-fibrotic acute stress response (Figure 5a). In the liver, which was included as a control, doxorubicin stimulated the expression of both pro-inflammatory and pro-fibrotic cytokines and lovastatin blocked both types of stress responses (Figure 5b).

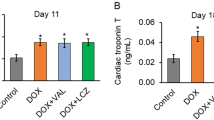
Lovastatin attenuates acute and subacute toxic effects of doxorubicin in vivo. (a and b) Balb/c mice were left untreated or were pre-treated twice with lovastatin (Lova) (10 mg/kg; p.o.). At 48 h after exposure to a single dose of doxorubicin (Doxo) (10 mg/kg; i.p.) mice were killed and the mRNA expression of pro-inflammatory (TNFα, IL-6) and pro-fibrotic (CTGF) genes was analyzed by real-time PCR in heart (a) and liver (b). Relative mRNA expression in the control was set to 1.0. (c) Balb/c mice were repeatedly exposed to doxorubicin (3 × 3 mg/kg; i.p.). Lovastatin was applied on a regular basis as described in methods. Mice were killed 21 days after the first doxorubicin administration. The mRNA expression of CTGF was analyzed in heart and set to 1.0 in untreated control. The gel picture shows the result of a semiquantitative RT-PCR analysis. The data shown in the histogram are abtained from quantitative real-time RT-PCR analyses as described in methods. (d) Balb/c mice were treated with doxorubicin and lovastatin as described under (c). Cardiac mRNA levels of ANP, BNP and Acta1 (d) were analyzed by quantitative real-time RT-PCR analysis using β-actin and GAPDH mRNA levels as internal controls. Relative mRNA levels in untreated controls was set to 1.0. (e) Cardiotoxicity was determined by analyzing the serum concentration of cardiac troponin I using ELISA-based method at day 9 and 16. Nd, not detectable. (f) Balb/c mice were treated with doxorubicin and lovastatin as described under C. Cardiac tissue sections were analyzed with regard to fibrosis using the Masson's trichrome staining protocol. Fibrotic tissue is stained in cyan color. Shown are representative pictures
In the clinic, the frequency and severity of congestive heart failure depends on the cumulative anthracycline dose and often occurs as a delayed effect. To mimic the clinical situation in humans, Balb/c mice were repeatedly treated with doxorubicin (3 × 3 mg/kg; i.p.) and subacute toxicity was monitored 21 days after the first doxorubicin administration. Lovastatin (10 mg/kg; p.o.) was administered 3–4 times a week. Doxorubicin caused loss in body weight of about 20%, which was not affected by lovastatin (see Supplementary Figure S2). The heart weight/body weight ratio was not significantly changed under these conditions (data not shown). Doxorubicin increased the mRNA levels of CTGF in the heart, which was largely impaired by the statin (Figure 5c). Lovastatin also mitigated the doxorubicin-provoked large increase in the amount of atrial natriuretic peptide (ANP) mRNA, as well as the concomitant downregulation of alpha skeletal muscle actin (acta1) mRNA expression (Figure 5d). mRNA level of the brain natriuretic peptide (BNP) was not altered under our experimental conditions (Figure 5d). Furthermore, lovastatin reduced the doxorubicin-stimulated increase in the serum level of cardiac troponin I (Figure 5e), showing that lovastatin counteracts subacute cardiotoxicity evoked by doxorubicin in vivo. This gains support by the finding that the onset of cardiac fibrosis, which was monitored by collagen staining (that is, Masson's trichrome staining) of cardiac tissue sections, was constrained by the statin (Figure 5f). Lovastatin did not show major effects on doxorubicin-stimulated acute changes in the expression of genes coding for antioxidative proteins or drug transporter (Supplementary Figure S3). Under situation of subacute doxorubicin exposure, lovastatin slightly mitigated the doxorubicin-induced increase in the amount of heme oxygenase-1 (ho-1) mRNA (Supplementary Figure S3). In addition, lovastatin lowered the doxorubicin-induced decrease in the number of peripheral leukocytes (data not shown). Collectively, a protective effect of lovastatin against the deleterious effects of anthracyclines was observed not only in vitro in H9c2 cardiomyoblasts, but also in vivo using a mouse model and therapeutically relevant doses.
Lovastatin affects doxorubicin-induced alterations in the gene expression of resistance-related factors
Genotoxic stress is known to provoke complex changes in gene expression, including players involved in DNA repair, checkpoint control and cell death that are major determinants of cellular sensitivity/resistance. Therefore, we examined the impact of lovastatin on doxorubicin-induced alterations in gene expression in the heart using the described subacute in vivo model. mRNA expression levels were analyzed by means of quantitative real-time RT-PCR making use of a semi-customised PCR array, which enables the analysis of the mRNA expression of 94 genes coding for major proteins involved in DNA repair, DNA damage response, checkpoint control and cell death. The screening analyses revealed quite complex changes in gene expression (see Supplementary Figure S4). Genes exhibiting considerable changes in their expression were selected for further validation by real-time RT-PCR analysis. As shown in Figure 6, doxorubicin caused upregulation of cell-cycle regulatory genes (cdc25A, wee1), as well as some DNA repair-associated genes (ddb2, rev1, brca2) and heme oxygenase-1 (ho-1). By contrast, the mRNA level of topo IIα was reduced following doxorubicin treatment (Figure 6). Lovastatin mitigated each of these stress responses. Some of the induced genes, such as ddb2, are induced by DNA damage.41, 42 Thus, these data strongly indicate that lovastatin suppresses DNA damage formation and downstream responses in the heart.

Lovastatin affects doxorubicin-induced changes in the mRNA expression of drug resistance-related factors. Balb/c mice were repeatedly exposed to doxorubicin (3 × 3 mg/kg; i.p.). Lovastatin was applied on a regular basis as described in methods. Mice were killed 21 days after the first doxorubicin administration. The mRNA level of genes involved in DNA repair, DNA damage response, cell-cycle progression and cell death was analyzed in heart using a semi-customized quantitative RT-PCR-array as described in methods. The results obtained (see Supplementary Figure S4) were confirmed by quantitative real-time RT-PCR for a subset of selected genes as depicted. The relative mRNA amount in untreated animals was set to 1.0 (dashed line). Changes in mRNA levels of ≥0.5 ≤ 2-fold (horizontal lines) were considered as not relevant
Lovastatin potentiates the antitumor activity of doxorubicin in vivo
From these data it is reasonable to conclude that lovastatin might reduce the patient's risk for heart failure following doxorubicin-based cancer therapy. However, does under these conditions the statin also mitigates the anticancer efficacy of doxorubicin? To address this concern, we examined the effect of lovastatin on doxorubicin-induced cell killing of human fibrosarcoma cells (HT-1080). Fibrosarcoma cells were chosen as the regimen for their treatment contains anthracyclines as first-line therapeutics. These data revealed that lovastatin did not protect HT-1080 cells against doxorubicin in vitro (Figures 7a and b). Rather, the statin provoked a weak sensitizing effect. Also, the doxorubicin-stimulated phosphorylation of H2AX was not reduced by lovastatin in HT-1080 cells (Figure 7c). To substantiate these data, we investigated the antitumor effect of doxorubicin and lovastatin on the growth of HT-1080 cells in vivo using a xenograft mouse model. Monotherapy with lovastatin caused only a very weak retardation, whereas administration of doxorubicin clearly reduced tumor growth (Figure 7d). Animals that received both doxorubicin and lovastatin showed a slower increase in tumor size as compared with untreated, lovastatin-treated or doxorubicin-treated individuals (Figure 7d). Taken together, co-treatment with lovastatin potentiates the antitumor efficacy of doxorubicin in a human fibrosarcoma xenograft model in vivo.

Effect of lovastatin on doxorubicin-induced responses of fibrosarcoma cells. Human HT-1080 fibrosarcoma cells were left untreated (−Lova) or were pre-treated with lovastatin (+Lova) (1 μM, overnight) before pulse treatment (1 h) with doxorubicin. (a and b) After incubation period of 72 h, cell viability was monitored (a). Relative cell viability in untreated cells was set to 100%. The percentage of apoptotic cells was analyzed by FACS-based determination of Annexin V positive cells (b). Results presented are the mean±S.D. from five independent experiments. (c) H2AX phosphorylation (γH2AX) was assayed 1 h after doxorubicin treatment by western blot (upper panel) or immunohistochemistry (lower panel). Doxo1 (0.2 μM); Doxo2 (0.3 μM) Doxo3 (1.0 μM). (d) For the xenograft study, HT-1080 fibrosarcoma cells were injected into both flanks of immunodeficient mice. After 5 days, lovastatin treatment started. Doxorubicin treatment was performed as described before. Tumor sizes were determined three times a week in each animal (number of tumors analyzed: Con, n=6; Lova, n=8; Doxo, n=6; Doxo +Lova, n=9). The lower part of the figure illustrates the tumor size 15 days after the onset of doxorubicin and/or lovastatin treatment
Discussion
Unwanted effects of anticancer drugs severely impact the prognosis and life quality of cancer patients. Therefore, the development of novel protective strategies preventing normal-tissue damage caused by anticancer drugs is of high clinical importance. Anthracycline derivatives such as doxorubicin are potent therapeutics. However, they provoke considerable acute and delayed normal tissue damage, in particular of the heart. Previously, we reported that the HMG-CoA reductase inhibitor lovastatin protects primary human endothelial cells from the deleterious effects of ionizing radiation and doxorubicin in vitro.27, 28 As cardiotoxicity is the clinically most relevant side effect of anthracyclines, we extended our in vitro studies to cardiomyoblasts. As shown here, lovastatin protects rat cardiomyoblasts from doxorubicin-induced cell death. This was related to attenuated DNA damage induction, as shown by a reduced Ser139 phosphorylation of histone H2AX, which is a generally accepted marker of the presence of DNA breaks.34, 35, 43 The geno-protective effect of lovastatin is independent of changes in doxorubicin transport and, furthermore, is not related to ROS, which are often discussed to trigger adverse effects of anthracyclines.44 Rather, it appears to rest upon a reduced inhibition of topo II by doxorubicin, resulting in mitigation of DNA damage formation. Hence, it is tempting to speculate that lovastatin interferes with either the formation or the processing to topo II cleavable complexes. Apparently, the low molecular weight GTPase Rac1 is essential for H2AX phosphorylation following doxorubicin exposure. This is consistent with the notion that Rho GTPases, in particular Rac1, are highly relevant targets of statins.19, 45, 46 These data are indicative of a so far not recognized crosstalk between Rho GTPases and genotoxic stress responses following the inhibition of topo II. The recent finding of a nuclear localization of Rac147 and Rho-activating GEFs48 support this hypothesis.
The usefulness of lovastatin as a cardioprotective drug was confirmed by measuring the acute and subacute toxicity of doxorubicin using an in vivo model system. Our data show that oral administration of lovastatin to Balb/c mice largely attenuated the acute pro–fibrotic stress response of cardiomyocytes (for example, the mRNA level of CTGF), which was observed 48 h after doxorubicin administration. A similar protective effect of lovastatin was found upon exposure of Balb/c mice to three consecutive doxorubicin doses and assaying subacute toxicity 8 days after the last doxorubicin treatment. Here, the mRNA amount of pro-fibrotic CTGF and of ANP, but not of BNP, were increased in response to doxorubicin while the the level of acta1 mRNA was reduced. Lovastatin was able to block these doxorubicin-stimulated alterations. Furthermore, lovastatin also alleviated cardiotoxicity as determined by the analysis of the serum concentrations of troponin I and by monitoring cardiac fibrosis in tissue sections. Changes in heart weight/body weight ratio were not observed after doxorubicin administration (data not shown). This might be due to the rather short postincubation times. The effect of doxorubicin and statins on chronic cardiac damage is subject of ongoing studies.
These data reported here show that lovastatin has a clear protective effect against doxorubicin-provoked heart damage. Regarding the mechanism possibly involved, we found that lovastatin mitigates doxorubicin-induced alterations in the gene expression of resistance-related factors, including the DNA repair protein DDB2 and cell-cycle regulatory phosphatases (that is, CDC25A, Wee1). As ddb2 is a DNA damage inducible gene,41, 42 these data indicate that lovastatin lowers the level of doxorubicin-induced cardiac DNA damage. This assumption is in line with our in vitro results obtained with cardiomyoblasts. Yet, we have to admit that, under our experimental conditions, we failed so far to detect doxorubicin-induced DNA damage in the heart in vivo. In our acute model system (that is, 48 h after administration of a single doxorubicin dose of 10 mg/kg), we could not detect an increase in cardiac H2AX phosphorylation, neither by immunohistochemistry nor by western blot analysis. Also after a shorter period of time (that is, 6 h after doxorubicin administration) western blot analysis failed to detect a significant change in γH2AX. Apparently, the degree of cardiac DNA damage induced in our experimental setting is quite low and, hence, difficult to detect. In this context it should be noted that in liver, which was included for reasons of control, doxorubicin was able to stimulate phosphorylation of H2AX (data not shown). As the statin also reduced the doxorubicin-induced drop in peripheral leukocyte count (data not shown) and alleviated acute hepatotoxicity, it appears possible that lovastatin counteracts multiple adverse pathways of anthracyclines in vivo. This hypothesis, which is currently under further investigation, gains support by previous reports showing that statins also reduce normal tissue damage, in particular of the intestine, following radiotherapy.29, 30, 31, 49 Moreover, statins are well known to protect the myocardium from acute ischemia-reperfusion injury32 and to alleviate aminoglycoside-induced nephrotoxicity.50 Collectively, our data together with published reports support the hypothesis of pleiotropic organoprotective functions of statins. Most important, we found that, while protecting cardiac tissue, lovastatin potentiated the killing efficacy of doxorubicin on cancer cells (that is, human fibrosarcoma cells) in vivo. This finding is in line with reports demonstrating an additive or synergistic effect of statins on anticancer drug- and radiation-induced tumor cell kill in vitro.19, 20
Overall, our in vitro and in vivo data provide evidence that lovastatin, which is a widely used lipid-lowering drug, protects heart cells against the acute and delayed side effects of the anthracycline derivative doxorubicin. At the same time, the statin promotes tumor cell death following doxorubicin treatment in vivo. The geno-protective effect of lovastatin on cardiomyocytes appears to be due to inhibition of Rac1-regulated functions and, furthermore, is related to the inhibition of topo II rather than ROS formation. In consequence, pro-fibrotic signaling mechanisms and cell death provoked by doxorubicin are reduced by lovastatin. Based on these data, we suggest that including lovastatin into current anthracycline-based tumortherapeutic protocols will widen the therapeutic window of anthracyclines by reducing cardiac damage and, at the same time, amending the tumor-cell response.
Materials and Methods
Materials
The following antibodies have been used: γH2AX anti-mouse antibody (pS139) (Millipore, Billerica, MA, USA), γH2AX anti-rabbit antibody (pS139) (Epitomics, Burlingame, CA, USA), ERK2 (Santa Cruz, CA, USA), Topo IIα (Enzo Life Sciences, Lörrach, Germany), Topo IIβ antibody (Abcam, Cambridge, MA, USA), p-Chk1 (Ser 345), Chk1 (Cell Signaling Technology, Inc., Beverly, MA, USA), peroxidise-conjugated secondary antibodies (Rockland, Gilbertsville, PA, USA) and Alexa Fluor 488 labeled secondary antibodies from Invitrogen (Paisley, UK). Alexa Fluor 488-conjugated Annexin V was from Axxora (Lörrach, Germany), WST-1 from Roche (Mannheim, Germany). Prenyltransferase inhibitor GGTI-286 and FTI-2148, the Rac1 inhibitor NSC23766, JNK inhibitor type II and p38 kinase inhibitor SB203580 were from Calbiochem (San Diego, CA, USA). RNeasy Mini Kit for total mRNA preparation and OmniScript Kit for cDNA synthesis were from Qiagen (Hilden, Germany), DNA oligos from SIGMA-Aldrich (Steinheim, Germany). The Mouse Cardiac Troponin I ELISA Kit was provided by Life Diagnostics (West Chester, PA, USA). Rho inactivating clostridial toxins Clostridium difficile toxin BF and Clostridium sordellii lethal toxin were generously provided by I Just and H Genth (Hannover, Germany).
Cell culture
H9c2 rat cardiomyoblasts were grown at 37 °C in DMEM supplemented with 20% fetal calf serum and penicillin/streptomycin. HT1080 cells were grown in RPMI supplemented with 5% fetal calf serum and penicillin/streptomycin.
Determination of viability and apoptotic cell death
Cell viability was monitored by use of the WST assay according to the manufacturers protocol (Roche Diagnostics, Mannheim, Germany). Apoptotic cell death was determined by analysing the fraction of FITC-Annexin V positive cells by FACS (Becton Dickinson, Heidelberg, Germany). Values given are the mean±S.E.M. of four to five independent experiments.
Cell-cycle analysis
At 24 h after treatment as indicated, cells were harvested and fixed in an ice-cold 70% ethanol. After RNase treatment (1 h, RT), DNA was stained by propidium iodide. Cell-cycle distribution was analyzed by FACS. Data shown are mean values from three independent experiments.
Western blot analysis
Total cell extracts were prepared by lysing identical number of cells (about 2–5 × 105) in Roti-load (Roth, Karlsruhe, Germany) sample buffer. Proteins were separated by SDS-PAGE (10–15% gels) and transferred onto nitrocellulose membranes. Membranes were then blocked in 5% non-fat milk in TBS/0.1% Tween 20 for ≥1 h at room temperature. Incubation with the primary antibody (as indicated) was conducted overnight at 4 °C. Incubation with peroxidase-conjugated anti-mouse or anti-rabbit secondary antibody (1 : 2000) (Rockland) was performed for 2 h at room temperature. Bound antibodies were then visualized by chemiluminescence and Hyperfilm ECL (GE Healthcare, Munich, Germany). Densitometrical quantification was performed with the Multi Analyst software (BioRad, Munich, Germany). Quantitative data from in vitro experiments are based on three independent experiments, in vivo data are calculated from the analysis of five to six mice.
Immunhistochemical analysis of H2AX phosphorylation
Cells were seeded onto cover slides. After treatment, cells were fixed with 4% paraformaldehyde (15 min, RT), followed by incubation with ice-cold methanol (−20 °C, 1 h). After blocking (PBS containing 0.1% TWEEN-20/1% BSA (w/v), 2 h, RT) incubation with an antibody specific for phosphorylated (Ser139), histone H2AX (1 : 500) was conducted overnight at 4 °C. Incubation with the secondary fluorescent-labeled antibody (Alexa Fluor 488; Invitrogen) was performed for 2 h at room temperature in the dark. For microscopical analysis a Zeiss Axiovert 35 (Carl Zeiss AG, Oberkochen, Germany) was used. Values given are the mean±S.D. from three independent experiments with 50 cells each being analyzed.
Analysis of DNA strand break induction
Formation of DNA strand breaks was assayed by the comet assay.51 Comets were visualized by microscopy and quantified by determination of the percentage of DNA in the tail (Komet 4.02, Kinetics Imaging, Merseyside, UK). A total of 50 nuclei were evaluated per treatment. The mean±S.D. from at least three independent experiments is shown.
Determination of doxorubicin transport (uptake and efflux)
To this end, doxorubicin-treated cells were fixed in an ice-cold 70% ethanol (−20 °C, 1 h) and analyzed by FACS (Becton Dickinson). The inherent fluorescence of doxorubicin (exc.: 470 nm; emm.: 575 nm) was taken as indication of the doxorubicin content of the cells. To measure doxorubicin uptake, cells were treated with doxorubicin for 1 h before FACS analysis was performed. To determine doxorubicin efflux, cells were pre-loaded with doxorubicin (1 h pulse treatment), followed by postincubation period of 6 h in the absence of the drug. The decrease in fluorescence occurring within this period of time is taken as indication of drug efflux. A representative histogram out of three independent experiments is shown.
In vivo experiments
Balb/c mice were 3–4 months of age and weighed an average of 20–25 g at the start of the experiments. In all, 24 (or 32 in case of the xenograft study) animals were randomly divided into groups of six animals per group (or eight animals per group in case of the xenograft study). To determine the effect of lovastatin on acute toxicity of doxorubicin, mice were pre-treated twice with lovastatin (10 mg/kg; p.o.). At 2 days after doxorubicin administration (10 mg/kg; i.p.) analyses were performed. To determine subacute toxicity of doxorubicin, animals were treated with lovastatin 3–4 times per week (10 mg/kg; p.o.). Three single doses of doxorubicin (3 mg/kg; i.p.) were applied on days 3, 9 and 16. At 8 days after the last doxorubicin injection, mice were killed. Body and heart weight was determined for the calculation of heart weight/body weight ratio. Organs were immediately frozen in liquid nitrogen and stored at −80 °C or were fixed in formalin and embedded in paraffin for histological analysis. Blood samples were collected at days 9 and 16 from the tail vein and at the end of the experiment from tail vein and heart. For the xenograft study, 4 × 106 HT1080 cells were injected subcutaneously into each flank of immunodeficient Rag2−/− mice. Tumors growing during the first week of the treatment were included in the final evaluation. Data shown are the mean±S.E.M.
Analysis of gene expression by PCR array
Balb/c mice were treated with doxorubicin (3 × 3 mg/kg, i.p) and lovastatin (3 × 10 mg/kg per week, p.o.) as described before. At 3 weeks after the first doxorubicin administration, the effect of lovastatin on doxorubicin-induced alterations in mRNA expression levels was analyzed. To this end, total RNA was isolated from heart for real-time PCR analysis. Real-time PCR analysis was performed using pooled mRNA samples from n=6 mice. The PCR array used in this study was semicustomised (established in collaboration with Sigma-Aldrich Chemie GmbH, Steinheim, Germany). It contains 94 genes involved in DNA repair, stress signaling, cell-cycle regulation and metastasis. Amplification was performed using the MyIQ real-time PCR cycler (BioRad). At the end of the run, melting curves were analyzed to ensure the specificity of the amplification reaction. PCR products with crossing points of ≥35 cycles were omitted. mRNA expression levels were normalized to that of the housekeeping genes GAPDH and β-actin. Relative gene expression in untreated control animals was set to 1.0 and compared with that of lovastatin-treated animals. Only changes in gene expression of ≤0.5 and ≥2-fold were considered as relevant. Selected genes that responded to doxorubicin or lovastatin treatment with relevant changes in mRNA expression were validated by performing additional real-time RT-PCR analyses (triplicate determinations).
Gene expression analyses (real-time RT-PCR)
Total RNA was purified from 20–30 mg of tissue using the RNeasy Mini Kit (Qiagen). The RT reaction was performed using the OmniScript Kit (Qiagen) using 500–2000 ng of mRNA. For each PCR reaction, 2 μl of diluted (1 : 10) cDNA, primers (0.42 μM each), and dNTPs (0.35 μM each; Perkin Elmer, Waltham, MA, USA) were used. PCR runs were carried out as follows: 1. 95 °C, 2 min; 2. 95 °C, 30 s–60 °C, 30 s–72 °C, 40 s; 3. 72 °C, 10 min. For semiquantitative end-point analysis, PCR products were separated on agarose gels and stained with ethidium bromide. Real-time RT-PCR analysis was performed with pooled RNA samples from up to six mice and was carried out in duplicates using the QPCR SYBR Green Fluorescein Mix (Thermo Fisher, Dreieich, Germany) and a MyIQ Thermal Cycler (BioRad). The following primers were used: GAPDH, f: 5′-AACTTTGGCATTGTGGAAGG-3′, r: 5′-CACATTGGGGGTAGGAACAC-3′ (222 bp product); β-actin, f: 5′-GCATTGCTGACAGGATGCAG-3′, r: 5′-CCTGCTTGCTGATCCACATC-3′ (159 bp); connective tissue growth factor (CTGF), f: 5′-CAAAGCAGCTGCAAATACCA-3′, r: 5′-GGCCAAATGTGTCTTCCAGT-3′ (220 bp); interleukin-6 (IL-6), f: 5′-AGTTGCCTTCTTGGGACTGA-3′, r: 5′-CAGAATTGCCATTGCACAAC-3′ (191 bp); tumor necrosis factor alpha (TNFα, f: 5′-AGCCCCCAGTCTGTATCCTT-3′, r: 5′-CTCCCTTTGCAGAACTCAGG-3′ (212 bp); heme oxygenase (ho-1); f: 5′-CACGCATATACCCGCTACCT-3′ r: 5′-CCAGAGTGTTCATTCGAGCA-3′; glutathione peroxidase (gpx-1), f: 5′-GTCCACCGTGTATGCCTTCT-3′ r: 5′-GAACTGATTGCACGGGAAAC-3′; multidrug resistance gene 1 (mdr-1), f: 5′-ACCATGGAGGAAATCACAGC-3′ r: 5′-TGGTGGCATCATCCAAGATA-3′; multidrug resistance-associated protein 1 (mrp-1): f: 5′-AGGCCTACTACCCCAGCATT-3′ r: 5′-CAGTCTCTCCACTGCCACAA-3′; atrial natriuretic peptide (ANP): f: 5′-CCTAAGCCCTTGTGGTGTGT-3′ r: 5′-CAGAGTGGGAGAGGCAAGAC-3′; brain natriuretic peptide (BNP): f: 5′-CTGAAGGTGCTGTCCCAGAT-3′ r: 5′-CCTTGGTCCTTCAAGAGCTG-3′; alpha sceletal muscle actin (acta1): f: 5′-GCGCAAGTACTCAGTGTGGA-3′ r: 5′-TCGTCCTGAGGAGAGAGAGC-3′. mRNA levels of GAPDH and β-actin were taken for normalization.
Determination of troponin I serum level and white blood cell count
Serum troponin I levels were detected using the Mouse Cardiac Tn-I ELISA (Life Diagnostics, West Chester, PA, USA). Values given are the mean+S.D. from at least three animals per group. White blood cell count was determined by routine analysis in the Institute of Clinical Chemistry and Laboratory Medicine (Universitätsmedizin Mainz, Germany). Data shown are based on the analysis of four to six animals per group.
Fibrosis staining
To detect fibrosis in cardiac tissue histologically, heart tissue was fixed with formalin and embedded into paraffin blocks by routine procedure (Institute of Pathology, Mainz). Three sections per heart were prepared for fibrosis staining using the Masson-Goldner trichrome staining kit (MERCK Darmstadt, Germany). Four animals per group were analyzed. Photographs were taken on a Zeiss Axiovert 35 microscope and representative pictures are shown.
Statistical analysis
For statistical analysis the Student's t-test or the Mann–Whitney U-test were applied. P–values of P≤0.05 were considered significant and marked with an asterisk.
Abbreviations
- Acta1:
-
alpha sceletal muscle actin 1
- ANP:
-
atrial natriuretic peptide
- β-actin:
-
beta actin
- BNP:
-
brain natriuretic peptide
- Brca2:
-
breast cancer susceptibility protein 2
- Cdc25a:
-
cell division cycle 25 homolog A
- Chk-1:
-
checkpoint kinase-1
- CTGF:
-
connective tissue growth factor
- DAPI:
-
4′,6-diamidino-2-phenylindole
- DCFDA:
-
dichlorofluorescein diacetate
- Ddb2:
-
DNA damage binding protein 2
- DDR:
-
DNA damage response
- Doxo:
-
doxorubicin
- DSB:
-
DNA double strand break
- ERK2:
-
extracellular signal-regulated kinase 2
- Eto:
-
etoposide
- FACS:
-
fluorescence-activated cell sorting
- FITC:
-
fluorescein isothiocyanate
- FTI:
-
farnesyl transferase inhibitor
- GAPDH:
-
glyceraldehyde 3-phosphate dehydrogenase
- GEFs:
-
guanine nucleotide exchange factors
- GGTI:
-
geranylgeranyl transferase inhibitor
- γH2AX:
-
Ser139 phosphorylated histone H2AX
- gpx–1:
-
glutathione peroxidase 1
- HMG-CoA:
-
3–hydroxy–3–methyl–glutaryl–coenzyme A
- ho–1:
-
heme oxygenase 1
- IL-6:
-
interleukin 6
- IR:
-
ionizing radiation
- Lova:
-
lovastatin
- mdr-1:
-
multidrug resistance protein 1
- mrp-1:
-
multidrug resistance-related protein 1
- NF-κB:
-
nuclear factor kappaB
- Rac1:
-
Ras-related C3 botulinum toxin substrate 1
- REV1:
-
human Rev1 homolog
- Rho:
-
Ras-homologous
- ROS:
-
reactive oxygen species
- RT-PCR:
-
reverse transcriptase-polymerase chain reaction
- SAPK/JNK:
-
stress-activated protein kinase/c-Jun-N-terminal kinase
- TcdBF:
-
Clostridium difficile toxin BF
- TcsL:
-
Clostridium sordellii lethal toxin
- TNFα:
-
tumor necrosis factor alpha
- topoII:
-
topoisomerase II
- Wee1:
-
human Wee1-like protein kinase
- WST-1:
-
water soluble tetrazolium
- XPA:
-
xeroderma pigmentosum complementation group A
- XPC:
-
xeroderma pigmentosum complementation group C
- XRCC1:
-
X-ray repair cross-complementing protein 1
References
Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L . Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev 2004; 56: 185–229.
Kremer LC, van Dalen EC, Offringa M, Ottenkamp J, Voute PA . Anthracycline-induced clinical heart failure in a cohort of 607 children: long-term follow-up study. J Clin Oncol 2001; 19: 191–196.
van Dalen EC, van der Pal HJ, Kok WE, Caron HN, Kremer LC . Clinical heart failure in a cohort of children treated with anthracyclines: a long-term follow-up study. Eur J Cancer 2006; 42: 3191–3198.
Ferreira AL, Matsubara LS, Matsubara BB . Anthracycline-induced cardiotoxicity. Cardiovasc Hematol Agents Med Chem 2008; 6: 278–281.
Lyu YL, Kerrigan JE, Lin CP, Azarova AM, Tsai YC, Ban Y et al. Topoisomerase IIbeta mediated DNA double-strand breaks: implications in doxorubicin cardiotoxicity and prevention by dexrazoxane. Cancer Res 2007; 67: 8839–8846.
Fogli S, Nieri P, Breschi MC . The role of nitric oxide in anthracycline toxicity and prospects for pharmacologic prevention of cardiac damage. Faseb J 2004; 18: 664–675.
Hasinoff BB, Herman EH . Dexrazoxane: how it works in cardiac and tumor cells. Is it a prodrug or is it a drug? Cardiovasc Toxicol 2007; 7: 140–144.
Yan T, Deng S, Metzger A, Godtel-Armbrust U, Porter AC, Wojnowski L . Topoisomerase II{alpha}-dependent and -independent apoptotic effects of dexrazoxane an. Mol Cancer Ther 2009; 8: 1075–1085.
Simunek T, Sterba M, Popelova O, Adamcova M, Hrdina R, Gersl V . Anthracycline-induced cardiotoxicity: overview of studies examining the roles of oxidative stress and free cellular iron. Pharmacol Rep 2009; 61: 154–171.
Marty M, Espie M, Llombart A, Monnier A, Rapoport BL, Stahalova V . Multicenter randomized phase III study of the cardioprotective effect of dexrazoxane (Cardioxane) in advanced/metastatic breast cancer patients treated with anthracycline-based chemotherapy. Ann Oncol 2006; 17: 614–622.
Hofland KF, Thougaard AV, Sehested M, Jensen PB . Dexrazoxane protects against myelosuppression from the DNA cleavage-enhancing drugs etoposide and daunorubicin but not doxorubicin. Clin Cancer Res 2005; 11: 3915–3924.
Weinstein DM, Mihm MJ, Bauer JA . Cardiac peroxynitrite formation and left ventricular dysfunction following doxorubicin treatment in mice. J Pharmacol Exp Ther 2000; 294: 396–401.
Cole MP, Chaiswing L, Oberley TD, Edelmann SE, Piascik MT, Lin SM et al. The protective roles of nitric oxide and superoxide dismutase in adriamycin-induced cardiotoxicity. Cardiovasc Res 2006; 69: 186–197.
Chaiswing L, Cole MP, Ittarat W, Szweda LI, St Clair DK, Oberley TD . Manganese superoxide dismutase and inducible nitric oxide synthase modify early oxidative events in acute adriamycin-induced mitochondrial toxicity. Mol Cancer Ther 2005; 4: 1056–1064.
Pacher P, Liaudet L, Bai P, Mabley JG, Kaminski PM, Virag L et al. Potent metalloporphyrin peroxynitrite decomposition catalyst protects against the development of doxorubicin-induced cardiac dysfunction. Circulation 2003; 107: 896–904.
Gianni L, Herman EH, Lipshultz SE, Minotti G, Sarvazyan N, Sawyer DB . Anthracycline cardiotoxicity: from bench to bedside. J Clin Oncol 2008; 26: 3777–3784.
Zhou Q, Liao JK . Pleiotropic effects of statins – Basic research and clinical perspectives. Circ J 2010; 74: 818–826.
Liao JK, Laufs U . Pleiotropic effects of statins. Annu Rev Pharmacol Toxicol 2005; 45: 89–118.
Fritz G . Targeting the mevalonate pathway for improved anticancer therapy. Curr Cancer Drug Targets 2009; 9: 626–638.
Cafforio P, Dammacco F, Gernone A, Silvestris F . Statins activate the mitochondrial pathway of apoptosis in human lymphoblasts and myeloma cells. Carcinogenesis 2005; 26: 883–891.
Agarwal B, Bhendwal S, Halmos B, Moss SF, Ramey WG, Holt PR . Lovastatin augments apoptosis induced by chemotherapeutic agents in colon cancer cells. Clin Cancer Res 1999; 5: 2223–2229.
Rao S, Lowe M, Herliczek TW, Keyomarsi K . Lovastatin mediated G1 arrest in normal and tumor breast cells is through inhibition of CDK2 activity and redistribution of p21 and p27, independent of p53. Oncogene 1998; 17: 2393–2402.
Feleszko W, Mlynarczuk I, Balkowiec-Iskra EZ, Czajka A, Switaj T, Stoklosa T et al. Lovastatin potentiates antitumor activity and attenuates cardiotoxicity of doxorubicin in three tumor models in mice. Clin Cancer Res 2000; 6: 2044–2052.
Dimitroulakos J, Ye LY, Benzaquen M, Moore MJ, Kamel-Reid S, Freedman MH et al. Differential sensitivity of various pediatric cancers and squamous cell carcinomas to lovastatin-induced apoptosis: therapeutic implications. Clin Cancer Res 2001; 7: 158–167.
Ahn KS, Sethi G, Aggarwal BB . Simvastatin potentiates TNF-alpha-induced apoptosis through the down-regulation of NF-kappaB-dependent antiapoptotic gene products: role of IkappaBalpha kinase and TGF-beta-activated kinase-1. J Immunol 2007; 178: 2507–2516.
Ahn KS, Sethi G, Aggarwal BB . Reversal of chemoresistance and enhancement of apoptosis by statins through down-regulation of the NF-kappaB pathway. Biochem Pharmacol 2008; 75: 907–913.
Damrot J, Nuebel T, Epe B, Roos WP, Kaina B, Fritz G . Lovastatin protects human endothelial cells from the genotoxic and cytotoxic effects of the anticancer drugs doxorubicin and etoposide. Br J Pharmacol 2006; 149: 988–997.
Nuebel T, Damrot J, Roos WP, Kaina B, Fritz G . Lovastatin protects human endothelial cells from killing by ionizing radiation without impairing induction and repair of DNA double-strand breaks. Clin Cancer Res 2006; 12: 933–939.
Ostrau C, Hulsenbeck J, Herzog M, Schad A, Torzewski M, Lackner KJ et al. Lovastatin attenuates ionizing radiation-induced normal tissue damage in vivo. Radiother Oncol 2009; 92: 492–499.
Haydont V, Bourgier C, Pocard M, Lusinchi A, Aigueperse J, Mathe D et al. Pravastatin Inhibits the Rho/CCN2/extracellular matrix cascade in human fibrosis explants and improves radiation-induced intestinal fibrosis in rats. Clin Cancer Res 2007; 13: 5331–5340.
Wang J, Boerma M, Fu Q, Kulkarni A, Fink LM, Hauer-Jensen M . Simvastatin ameliorates radiation enteropathy development after localized, fractionated irradiation by a protein C-independent mechanism. Int J Radiat Oncol Biol Phys 2007; 68: 1483–1490.
Ludman A, Venugopal V, Yellon DM, Hausenloy DJ . Statins and cardioprotection–more than just lipid lowering? Pharmacol Ther 2009; 122: 30–43.
Tewey KM, Rowe TC, Yang L, Halligan BD, Liu LF . Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase II. Science 1984; 226: 466–468.
Harper JW, Elledge SJ . The DNA damage response: ten years after. Mol Cell 2007; 28: 739–745.
Olive PL . Detection of DNA damage in individual cells by analysis of histone H2AX phosphorylation. Methods Cell Biol 2004; 75: 355–373.
Rothkamm K, Lobrich M . Evidence for a lack of DNA double-strand break repair in human cells exposed to very low x-ray doses. Proc Natl Acad Sci USA 2003; 100: 5057–5062.
Stiff T, O’Driscoll M, Rief N, Iwabuchi K, Lobrich M, Jeggo PA . ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res 2004; 64: 2390–2396.
Nitiss JL . DNA topoisomerases in cancer chemotherapy: using enzymes to generate selective DNA damage. Curr Opin Investig Drugs 2002; 3: 1512–1516.
Huelsenbeck J, Dreger S, Gerhard R, Barth H, Just I, Genth H . Difference in the cytotoxic effects of toxin B from Clostridium difficile strain VPI 10463 and toxin B from variant Clostridium difficile strain 1470. Infect Immun 2007; 75: 801–809.
Huelsenbeck J, Dreger SC, Gerhard R, Fritz G, Just I, Genth H . Upregulation of the immediate early gene product RhoB by exoenzyme C3 from Clostridium limosum and toxin B from Clostridium difficile. Biochemistry 2007; 46: 4923–4931.
Batista LF, Roos WP, Christmann M, Menck CF, Kaina B . Differential sensitivity of malignant glioma cells to methylating and chloroethylating anticancer drugs: p53 determines the switch by regulating xpc, ddb2, and DNA double-strand breaks. Cancer Res 2007; 67: 11886–11895.
Takimoto R, MacLachlan TK, Dicker DT, Niitsu Y, Mori T, el-Deiry WS . BRCA1 transcriptionally regulates damaged DNA binding protein (DDB2) in the DNA repair response following UV-irradiation. Cancer Biol Ther 2002; 1: 177–186.
Rothkamm K, Kruger I, Thompson LH, Lobrich M . Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol Cell Biol 2003; 23: 5706–5715.
Menna P, Salvatorelli E, Minotti G . Anthracycline Degradation in Cardiomyocytes: A Journey to Oxidative Survival. Chem Res Toxicol 2009; 23: 6–10.
Wang CY, Liu PY, Liao JK . Pleiotropic effects of statin therapy: molecular mechanisms and clinical results. Trends Mol Med 2008; 14: 37–44.
Rashid M, Tawara S, Fukumoto Y, Seto M, Yano K, Shimokawa H . Importance of Rac1 signaling pathway inhibition in the pleiotropic effects of HMG-CoA reductase inhibitors. Circ J 2009; 73: 361–370.
Sandrock K, Bielek H, Schradi K, Schmidt G, Klugbauer N . The nuclear import of the small GTPase Rac1 is mediated by the direct interaction with karyopherin alpha2. Traffic 2010; 11: 198–209.
Guerra L, Carr HS, Richter-Dahlfors A, Masucci MG, Thelestam M, Frost JA et al. A bacterial cytotoxin identifies the RhoA exchange factor Net1 as a key effector in the response to DNA damage. PLoS One 2008; 3: e2254.
Monceau V, Pasinetti N, Schupp C, Pouzoulet F, Opolon P, Vozenin MC . Modulation of the Rho/ROCK Pathway in Heart and Lung after Thorax Irradiation Reveals Targets to Improve Normal Tissue Toxicity. Curr Drug Targets 2010; 11: 1395–1404.
Antoine DJ, Srivastava A, Pirmohamed M, Park BK . Statins inhibit aminoglycoside accumulation and cytotoxicity to renal proximal tubule cells. Biochem Pharmacol 2010; 79: 647–654.
Olive PL, Banath JP . The comet assay: a method to measure DNA damage in individual cells. Nat Protoc 2006; 1: 23–29.
Acknowledgements
We would like to thank Katrin Roth, Rebekka Kitzinger and Adriana Degreif for excellent technical support. This work was supported by a grant from Deutsche Krebshilfe (107361).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by A Stephanou
Supplementary Information accompanies the paper on Cell Death and Disease website
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Huelsenbeck, J., Henninger, C., Schad, A. et al. Inhibition of Rac1 signaling by lovastatin protects against anthracycline-induced cardiac toxicity. Cell Death Dis 2, e190 (2011). https://doi.org/10.1038/cddis.2011.65
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2011.65
Keywords
This article is cited by
-
Endothelial-to-Mesenchymal Transition: Potential Target of Doxorubicin-Induced Cardiotoxicity
American Journal of Cardiovascular Drugs (2023)
-
Statins for Cardiac and Vascular Protection During and After Cancer Therapy
Current Oncology Reports (2022)
-
Doxorubicin-Induced Cardiotoxicity: An Overview on Pre-clinical Therapeutic Approaches
Cardiovascular Toxicology (2022)
-
Rac1-mediated cardiac damage causes diastolic dysfunction in a mouse model of subacute doxorubicin-induced cardiotoxicity
Archives of Toxicology (2018)
-
Rho inhibition by lovastatin affects apoptosis and DSB repair of primary human lung cells in vitro and lung tissue in vivo following fractionated irradiation
Cell Death & Disease (2017)

{kind=link}
{kind=link}
{kind=link}
{kind=link}