
Abstract
During early neurogenesis, retinal neuronal cells display a conserved differentiation program in vertebrates. Previous studies established that nitric oxide (NO) and cGMP accumulation regulate essential events in retinal physiology. Here we used pharmacological and genetic loss-of-function to investigate the effects of NO and its downstream signaling pathway in the survival of developing avian retinal neurons in vitro and in vivo. Six-day-old (E6) chick retinal cells displayed increased calcium influx and produced higher amounts of NO when compared with E8 cells. L-arginine (substrate for NO biosynthesis) and S-nitroso-N-acetyl-D,L-penicillamine (SNAP; a nitrosothiol NO donor) promoted extensive cell death in E6 retinas, whereas in E8 both substances decreased apoptosis. The effect of NO at both periods was mediated by soluble guanylyl cyclase (sGC) and cGMP-dependent kinase (cGK) activation. In addition, shRNA-mediated cGKII knockdown prevented NO-induced cell death (E6) and cell survival (E8). This, NO-induced cell death or cell survival was not correlated with an early inhibition of retinal cell proliferation. E6 cells also responded differentially from E8 neurons regarding cyclic AMP-responsive element-binding protein (CREB) activation in the retina in vivo. NO strongly decreased nuclear phospho-CREB staining in E6 but it robustly enhanced CREB phosphorylation in the nuclei of E8 neurons, an effect that was completely abrogated by cGKII shRNAs at both embryonic stages. The ability of NO in regulating CREB differentially during retinal development relied on the capacity of cGKII in decreasing (E6) or increasing (E8) nuclear AKT (V-Akt murine thymoma viral oncogene) activation. Accordingly, inhibiting AKT prevented both cGKII shRNA-mediated CREB upregulation in E6 and SNAP-induced CREB activation in E8. Furthermore, shRNA-mediated in vivo cGKII or in vitro CREB1 knockdown confirmed that NO/cGKII dualistically regulated the downstream CREB1 pathway and caspase activation in the chick retina to modulate neuronal viability. These data demonstrate that NO-mediated cGKII signaling may function to control the viability of neuronal cells during early retinal development via AKT/CREB1 activity.
Similar content being viewed by others


Main
The retina is part of the central nervous system (CNS) because of its derivation from the anterior neural tube. Mature retinal tissue is arranged in a laminar structure composed of seven major classes of cells: ganglion, horizontal, amacrine, bipolar, cone and rod photoreceptors and the Müller glia.1 Early retinal tissue is a pseudo-stratified epithelium in which mitotically active neuronal precursor cells are found. Another noteworthy characteristic of vertebrate retinogenesis is the well-established chronology of cell-type generation, with ganglion cells generally being the first cells to be born and the Müller glia and bipolar neurons usually the last. Nevertheless, the differentiation periods of different cell types most frequently correlate and overlap with one another during both embryonic and postnatal periods.2
The gaseous signaling molecule nitric oxide (NO) is produced from the amino-acid L-arginine in a reaction catalyzed by the enzyme NO synthase (NOS). The reaction requires molecular oxygen, which produces NO and L-citrulline concomitantly.3 NO can mediate its effects basically by two mechanisms: first, the classical activation of soluble guanylyl cyclase (sGC) and cGMP-dependent kinases (cGKs), known as the NO/sGC/cGK pathway4, 5; second, NO can covalently attach to cysteine residues within the amino-acid sequence of specific proteins, a mechanism known as S-nitrosylation.6, 7 The involvement of NO and its related downstream signaling pathways have been previously demonstrated to control various CNS functions, including neural proliferation, survival and differentiation.8, 9, 10 We have previously demonstrated an anti-proliferative role for NO in the retina,11 and also a neuroprotective effect upon cultured retinal neurons.12 Moreover, there is a high-affinity transport system for L-arginine in cultured retinal neurons and an abundant expression of neuronal NOS (nNOS) in these cells.13 In such cultures, endogenous and exogenous NO modulate NF-kB activation and ascorbate transport through the canonical NO pathway.14 In addition, excitatory retinal cell death induced by α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors is regulated by nNOS activation.15 Therefore, NO regulates diverse signaling events in the retina.
Prominent roles of cGKs in mediating the actions of NO/cGMP system in different tissues have been upheld mainly using cGKI and cGKII knockout mice.16 For instance, cGKI or cGKII display pivotal roles in the cardiovascular system.17 Vascular smooth muscle tonus is regulated by an interaction between cGKIα and myosin phosphatase in the contraction apparatus of smooth muscle cells.18 Moreover, cGKI and cGKII are expressed in the CNS and regulate important neuronal functions. In hippocampal neurons, cGKI has been implicated in activity-dependent presynaptic potentiation19 and cGKII regulates AMPA receptors trafficking at the postsynaptic neuron.20 Pre- and postsynaptic actions of cGKs may involve a parallel activation of the small GTPase RhoA in the regulation of actin cytoskeleton, which contributes to synaptic strengthening in hippocampal neurons.21 In the developing retina, cGKs have been associated with Src avian sarcoma (Schmidt-Ruppin A-2) viral oncogene homolog (Src)-induced neuronal apoptosis,15 ascorbate transport14 and cGKII, specifically, regulates V-Akt murine thymoma viral oncogene (AKT)/PKB activation.22 In addition, cyclic AMP-responsive element-binding protein (CREB) phosphorylation in retinal cells is dependent on a cGK-mediated extracellular regulated kinase (ERK) activation, which directly correlates glutamate and NO pathway to nuclear CREB activity in the retina.23 Therefore, NO-mediated cGK activation also regulates physiological events in developing retinal cells.
CREB transcription factor belongs to the bZIP family of transcriptional regulators and has been associated with neuronal development.24 A critical serine residue within CREB KID domain (Ser133) controls its transcriptional activity via a calcium-dependent mechanism in neurons.25 CREB balances survival and death decisions in developing neuronal populations and its activity directly impacts on transcription of pro-survival genes such as B-cell CLL/lymphoma 2 (bcl-2) and brain-derived neurotrophic factor (bdnf).26 In retinal neurons, CREB phosphorylation was proposed as a mechanism for the neuroprotective actions of adenosine27 and fibroblast growth factor (FGF).28 Moreover, in cortical neurons, NO regulates CREB-dependent transcription by increasing CREB-DNA binding in CREB-responsive promoters.29
In the present work, we studied the modulatory effects of NO in the survival of developing chick retinal neurons in vitro and in vivo. We demonstrate that NO, through sGC and cGKII, differentially regulates the survival of retinal neurons, depending on the developmental period. As such, we propose that NO-induced neuronal cell death or survival was highly associated with the capacity of the NO/cGKII pathway either to downregulate or upregulate nuclear CREB1 activation in the retina.
Results
Considering the capacity of microenvironmental molecules to modulate/determine retinal neuronal cells development, and taking into account the neuromodulatory role played by NO in our culture system,11, 14, 15, 23 we tested whether prolonged NO treatment of cultured neurons would modulate the viability of those cells. S-nitroso-N-acetyl-D,L-penicillamine (SNAP; NO donor) effect was only detected when it was added 2 h after seeding E6 and E8 neurons, designed here as E6C0 or E8C0 (Figure 1). E6 cultures treated with SNAP in C0 (day of culturing) and observed in C4 (96 h after culturing) showed an extensive reduction in the number of viable neurons (Figures 1a and b), however, SNAP had no significant effect when added in C1 (24 h after culturing) in E6 cultures (−0.3±0.6%; data not shown). In E8 neuronal cultures, it was established that prolonged NO treatment had a neuroprotective effect in response to cytotoxic challenges.12, 30 For this reason, we compared the effects of SNAP in E8 purified neuronal cultures at C0 (Figure 1c). In contrast to E6 purified cultures, 4-day treatment with SNAP in E8 cultures increased the number of viable neurons. In order to characterize better the possible neurotoxic effect of NO in E6C0-C4 period, we used an exclusion assay with acridine orange (AO) and ethidium bromide (EB) to detect pyknotic cells. This protocol allowed us to confirm the reduction in the number of cells induced by SNAP in purified E6 cultures (Figure 1d upper panels and Figure 1e). Note that few cells were labeled with AO, and that increased condensed nuclei could be visualized in SNAP-treated cultures. Conversely, in E8C0-C4 (Figure 1d bottom panels and Figure 1f), pyknosis decreased when purified cultures were treated with SNAP. It is well known that glial cells are key factors in neuronal physiology, including in the retina. Hence, to evaluate the potential role of glial cells in modulating NO-dependent cell death or survival, we directly measured apoptosis in mixed retinal cultures. This mixed culture paradigm differs from purified neuronal cultures by the presence of Müller glial cells in a proportion of 15–20 to 80–85% neurons.14 Regardless Müller cells presence, in E6 mixed cultures, apoptotic cells (terminal deoxynucleotidyl transferase dUTP nick end labeling+, TUNEL+) increased in SNAP-treated conditions (Figure 1g), whereas the number of apoptotic cells decreased in SNAP-treated E8 mixed cultures (H), supporting the notion that NO directly regulates neuronal viability without the direct contribution of Müller cells. To validate our in vitro findings physiologically, we performed experiments in vivo, injecting SNAP in ovo. Interestingly, injection of SNAP in E6 embryos increased apoptotic labeling in the retina after 3 days (E9) (Figure 1i), whereas the same treatment reduced TUNEL+ cells in the retinas of embryos injected at E8 and analyzed at E11 (Figure 1j). Collectively, these findings suggest that NO regulates the in vivo viability of retinal neurons in a development-dependent manner.

NO dualistically modulates the survival of developing retinal neurons. Representative photomicrographs of E6 cultures (a; upper panels) or E8 cultures (a; bottom panels) cultivated for 96 h (C4). The histograms (b and c) represent neuronal cell survival by cell counting in E6 (b) or E8 purified cultures (c) after SNAP treatment (from day 0 (C0; day of culturing) up to day 4 (C4; 96 h after culturing)). Yellow arrow denotes a cluster of dead cells. Note the increased cell death in E6 cultures after SNAP treatment and the increased viability of neurons in E8 cultures. Calibration bars=10 μm. Total cell numbers in (b): CT=3300 cells, n=25; in (c): CT=1920 cells, n=20. (d–f), AO (green)/EB (red) exclusion assays. E6 purified cultures (upper panels in d and e) or E8 purified cultures (bottom panels in d and f) were treated with 100 μM SNAP from C0 up to C4 and then the assay was carried out. Note the decreased number of green cells and increased number of red cells (increased cell death index) in E6 and the opposite in E8 cultures. Calibration bars=10 μm. (g–h) NO regulates apoptosis in E6 and E8 mixed cells. TUNEL was carried out in E6 or E8 mixed cultures. Representative photomicrographs are displayed. Calibration bar=20 μm. Histograms represent TUNEL-positive neurons (green). (i and j) NO regulates apoptosis in vivo. E6 (i) or E8 (j) embryos were injected with SNAP and 3 days later (E9 or E11, respectively) retinas were evaluated by TUNEL. Arrowheads indicate apoptotic cells. Calibration bar=50 μm. (k) A representative cone photoreceptor (PTR; left photomicrograph) and a MN (right photomicrograph) from a purified retinal culture cultivated for 4 days. Note the distinctive features of both cell types in culture: cone photoreceptors can be distinguished by their characteristic lipid droplet and polarized morphology. (l and m) Cell population in E6 and E8 cultures affected by NO. PTRs and MNs number were assessed by morphological evaluation in E6 (l) or E8 (m) purified cultures. Data represent the mean±S.E.M. (error bars). *P<0.05, **P<0.01, ***P<0.001 in relation to control; N=5, t-test
We next evaluated the cellular population responsive to NO in E6 and E8 purified cultures. We used the criteria first adopted by Adler and Hatlee31 to discriminate between cone photoreceptors and multipolar neurons (MNs) in our culture paradigm. In this case, in chick retinal cultures, cone photoreceptors are very distinctive from MNs because of their characteristic lipid droplets, which make them easily distinguishable from MNs in culture (Figure 1k). Moreover, SNAP administration decreased the number of both photoreceptors and MNs in E6 cultures (Figure 1l). Significantly, SNAP only increased the viability of MNs in E8 cultures (Figure 1m). Therefore, in E6 cultures both photoreceptors and MNs are susceptible to SNAP-mediated cytotoxicity, whereas NO increases the survival of MNs in E8 cultures.
Next, the role of endogenous NO and constitutive activation of nNOS in regulating neuronal survival in E6 and E8 cultures was evaluated. Endogenous NO production was stimulated using L-arginine and constitutive nNOS activity was impaired using Nω-nitro-L-arginine methyl ester hydrochloride (L-NAME; a broad NOS inhibitor). Control experiments confirmed the expected increase in endogenous NO generation when cultures were incubated with L-arginine, whereas L-NAME treatment significantly decreased basal NO production in E6 and E8 purified cultures (Figures 2a and b, respectively). Furthermore, in E8 cultures, L-arginine treatment has already been described to increase endogenous NO production in an nNOS-dependent manner.14, 22 Interestingly, L-arginine administration promoted robust dose-dependent neuronal death in E6 purified cultures (Figure 2c) but it increased the viability of cultured E8 neurons (Figure 2e). Similarly, inhibiting basal NOS activity with L-NAME increased the viability of E6 neurons (Figure 2d) and promoted neuronal death in cultured E8 neurons (Figure 2f). To compare basal nNOS activity and endogenous NO production between E6 and E8, we measured calcium influx in cultured E6 and E8 neurons and direct NO production by 4-amino-5-methylamino-2',7'-difluorofluorescein diacetate (DAF-FM-DA) fluorescence in vivo. Radiolabeled calcium uptake was significantly higher in E6 cultured cells than in E8 neurons (Figure 2g). Moreover, endogenous NO production in E6 was also higher in relation to E8 retinas, as measured by DAF labeling (Figure 2h). These data suggest that a high calcium dynamics in E6 retinal neurons is probably related with the increased NO production in E6 relative to E8 retinas. To correlate the concentration of NO with neuronal death or survival, dose-response curves of SNAP were performed in E6 and E8 mixed cultures. In E6, SNAP was neurotoxic even in concentrations as low as 10 μM (Figure 2i, open circles), which supports the notion that NO in E6 may trigger a cell death pathway. However, in E8, SNAP displayed a biphasic cell survival curve, with concentrations up to 100 μM being neuroprotective and doses above 100 μM being neurotoxic (Figure 2i, black circles), suggesting that NO amounts are directly involved in promoting the dual effect observed in E6 and E8 cultures. Similar results were previously observed in E8 purified retinal neuronal cultures.12 To further validate this premise, another NO donor (Deta NONOate) was used. Indeed, low NONOate concentration also promoted extensive cell death in E6 mixed cultures (Figure 2j), while it significantly decreased cell death in E8 (Figure 2k).

Endogenous NOS activity and NO production regulate the survival of cultured retinal neurons. Endogenous NO production in cultured retinal neurons was regulated using L-arginine (L-Arg) and L-NAME and monitored by DAF-FM-DA labeling (a, E6 and b, E8). Calibration bar in a and b=10 μm, E6 (c and d) or E8 (e and f) cells were treated with L-arginine or L-NAME in C0, cultured up to C4, fixed and counted. Neuronal survival in (c–f) was evaluated by counting viable neurons in purified cultures. Total cell numbers in (a): CT=612 cells, n=6; in (b): CT=780 cells, n=6; in (c): CT=486 cells, n=6; in (d): CT=540 cells, n=6. Data represent the mean±S.E.M. (error bars). **P<0.01, ***P<0.001 in relation to control, one-way ANOVA. In g, E6 or E8 purified cultures were pulsed in C1 with radiolabeled calcium for 2 min. Cells were lysed and the homogenates were processed for liquid scintillation. In h, E6 or E8 retinas were incubated with DAF-FM-DA (5 μM) in HBSS for 30 min. Retinas were washed, flat-mounted and visualized in a confocal microscope. Calibration bars=50 μm. Histograms display the pixel count versus pixel intensity in E6 or E8 retinas labeled with DAF-FM-DA. (i) Dose-response curve of SNAP in E6 and E8 mixed cultures. Cell death was evaluated by EthD1 labeling. EC50 for SNAP in E6=68.4 μM. EC50 for SNAP in E8=148.2 μM. Total EthD1-positive cells in (i): CT E6=1456 cells, n=3. In j and k, DETA NONOate also regulates neuronal survival differentially. E6 (j) or E8 (k) mixed culture was treated with DETA NONOate (500 nM). EthD1 measured cell death. EthD1-positive cells: CT E6=237, n=3; CT E8=156, n=3. Data represent the mean±S.E.M. (error bars). *P<0.05 relative to CT in j and k, t-test
The classic intracellular pathway activated by NO was previously shown to depend on sGC activation, rise in intracellular cGMP levels and cGK modulation.32, 33 Consequently, we verified whether NO-induced neuronal cell damage or survival occurred through this pathway. Pharmacological activators of the canonical NO pathway such as 8Br-cGMP and Zaprinast (a cGMP phosphodiesterase inhibitor) or the sGC activator YC-1 promoted massive neuronal cell loss in E6 (Figure 3a) and increased neuronal survival in E8 cultures (Figure 3c). In addition, SNAP-induced cell death (Figure 3b) or survival (Figure 3d) was abrogated by 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ; a sGC inhibitor) or KT5823 (a cGK blocker). However, the thiol dithiothreitol (DTT), which blocks nitroxyl (NO−)-mediated cysteine modification (S-nitrosylation) but not NO (NO.)-dependent sGC activation,34, 35, 36 did not protect cultured cells from SNAP cytotoxic stimulus (59.5±9.5%, N=3, one-way analysis of variance (ANOVA)) in E6 purified retinal cultures, which argues against an S-nitrosylation-dependent mechanism for NO to increase death of E6 neurons. To corroborate our pharmacological data further, we used a lentiviral-mediated shRNA delivery system to knockdown cGKII specifically in retinal mixed cultures (Figure 3e) and evaluated SNAP-induced cell death or survival by using ethidium homodimer-1 (EthD1) labeling (Figure 3f) or 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Figures 3g and h). By these means, SNAP-induced neuronal death (E6; Figure 3f upper panels and 3g) or survival (E8; Figure 3f bottom panels and 3h) was completely abrogated in cells in which cGKII expression was knocked down. These data suggest that NO activates cGKII to selectively and dualistically regulate the viability of developing retinal neurons.

NO differentially regulates cultured retinal neuron viability in a cGKII-dependent manner. In a–d, Endogenous NO promotes neuronal death or survival. E6 (a and b) or E8 purified cultures (c and d) were treated in C0 with 100 μM SNAP, 5 μM ODQ, 800 nM KT5823, 25 μM zaprinast, 50 μM 8Br-cGMP or 10 μM YC-1 and quantified in C4. Cell survival, in graphs (a–d), was evaluated by counting of viable cells in purified cultures. Overall cell number in (a): CT=456 cells, n=4; in (b): CT=528 cells, n=4; in (c): CT=360 cells, n=4; in (d): CT=331 cells, n=4. Data represent the mean±S.E.M. (error bars). *P<0.05, **P<0.01, ***P<0.001 in relation to control, one-way ANOVA. In e, Validation of cGKII shRNA-induced cGKII knockdown in E6 and E8 mixed cultures. pLKO=empty vector (transduction control). In f–h, NO regulates retinal cell death or survival via cGKII. Cell death evaluation in mixed cultures by ethidium homodimer-1 (EthD1; purple) labeling (f) or MTT viability assay (g and h) was carried out in C4. Calibration bars=10 μm. EthD1-positive cells in f: pLKO E6=474 cells and pLKO E8=456 cells, n=3. Data represent the mean±S.E.M. (error bars). aP<0.05; aaaP<0.001 in relation to pLKO in F.2 and F.1, respectively. bP<0.05; bbbP<0.001 in relation to pLKO+SNAP in F.2 and F.1, respectively. ccP<0.05; cccP<0.001 in relation to pLKO+SNAP in F.2 and F.1, respectively; one-way ANOVA. *P<0.05; ***P<0.001 in relation to pLKO in h and g, respectively. #P<0.05, ##P<0.01, ###P<0.001 in relation to pLKO+SNAP in g and h; N=3, one-way ANOVA
We next evaluated the kinetics of the NO effect during in vitro development of E6 and E8 neurons. Cell survival data (Figure 4a) in purified cultures revealed that SNAP was toxic following prolonged in vitro incubation of E6 cultures. SNAP started to promote neurotoxicity in these cultures only after 96 h of administration (Figure 4a). Accordingly, the neuroprotective effect of SNAP in purified E8 cultures could only be observed after long-term incubation (Figure 4b). In order to establish a link between NO-induced cell death/survival and modulation of neuronal cell proliferation, we applied [3H]-thymidine pulses to cultured E6C0 or E8C0 neurons and challenged them with SNAP. SNAP treatment strongly reduced [3H]-thymidine incorporation in both E6 and E8 cultures (Figures 4c and d, respectively), an effect that was not abolished by ODQ, but was abrogated by DTT (Figures 4c and d). Therefore, cell death promoted by NO did not involve an earlier mitosis arrest of either E6 or E8 neurons.

Long-term SNAP treatment regulates the survival of retinal neurons. In a and b, NO-controlled neuronal death or survival is a long-term event. E6 (a) or E8 (b) purified cultures were treated in C0 with SNAP. Next, cultures were fixed and cell counting was performed in C1 (24 h after seeding), C2 (48 h after seeding), C3 (72 h after seeding) and C4 (96 h after seeding). In c and d, NO-mediated neuronal death or survival is independent of cell cycle arrest. E6 or E8 cells were treated with 100 μM SNAP, 5 μM ODQ or 50 μM DTT in C0, then cultures were pulsed with [3H]-thymidine (2 μCi/ml) for 1 h in C1. The material was analyzed by liquid scintillation. Data represent the mean±S.E.M. (error bars). ***P<0.001 in relation to control; ###P<0.001 in relation to SNAP; one-way ANOVA. Overall cell number in (a): CT=446 cells, n=4; in (b): CT=329 cells, n=4
The transcription factor CREB is primarily known for its neuroprotective role in neuronal cells.29, 37, 38, 39 We therefore addressed the role of NO in modulating CREB Ser133 phosphorylation (an index of CREB activation) in E6 and E8. The ontogeny curve of CREB in the developing retina indicated that CREB expression peaks in E6 and E14 (Figure 5a). Furthermore, CREB expression is higher in E6 than in E8 retina (Figure 5a), suggesting that CREB may be involved in controlling neuronal viability differentially in E6 and E8. Treatment of E6 mixed cultures with SNAP or YC-1 (a potent activator of sGC; Figure 5b, black bars) reduced phospho-CREB levels. Figure 5b (dashed bars) also shows that in E8 cultures, on the other hand, incubation with these compounds increased CREB phosphorylation. Significantly, as observed for NO-induced cell death, YC-1 or SNAP-induced phospho-CREB decrease in E6 cultures was blocked by KT5823, a cGK inhibitor (Figure 5c). In addition, in E8 cultures, the YC-1 or SNAP-induced phospho-CREB increase was also prevented by KT5823 (Figure 5d). The capacity of SNAP to induce phosphorylation of CREB in the retina in vivo was also evaluated. Interestingly, SNAP (injected at E6) downregulated CREB phosphorylation in E9 retinas, whereas it upregulated phospho-CREB in E11 retinas when injected at E8 (Figures 5e and f, respectively).

NO/cGK modulate CREB phosphorylation in retinal neurons. In a, developmental curve of CREB expression in the chick retina. In b–e, dual regulation of phospho-CREB by NO is cGK-dependent. E6 (black bars in b or c) or E8 (dashed bars in b or black bars in d and e) mixed cultures were treated in C0 with 100 μM SNAP (b–d) or 300 μM SNAP (e), 800 nM KT5823 or 10 μM YC-1 for 24 h. In (c), cultures were processed for western blotting or immunocytochemistry against phospho-CREB. Data represent the mean±S.E.M. (error bars). ***P<0.001 relative to control in b–d, **P<0.01 versus control in e; one-way ANOVA, N=3. In f and g, NO regulates CREB activation in the in vivo retina. Representative photomicrographs and densitometry of phospho-CREB labeling. Embryos were in ovo-injected with vehicle (sterile water; CT) or 100 μM SNAP in E6 (f) or E8 (g). Eggs were incubated up to E9 (f) or E11 (g). Calibration bar in f and g=50 μm. Data represent the mean±S.E.M. (error bars). **P<0.01 in relation to control in f and g; t-test, N=6
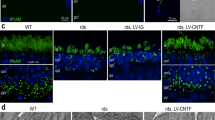
We next focused in the specific role played by cGKII in NO-regulated CREB function. Double-labeling immunocytochemistry demonstrated that cGKII and CREB were colocalized in cultured neurons (Figure 6a.1) and in vivo (Figure 6a.2) in E6 and E8. We next used a quantitative confocal analysis to study the role of the NO/cGKII pathway in regulating phospho-CREB within the nucleus. Confocal data showed that SNAP decreased (E6; Figure 6b) or increased (E8; Figure 6c) phospho-CREB/DAPI colocalization in puncta in pLKO cells (control). However, in E6 and E8 cultures in which cGKII was knocked down, the SNAP effect in regulating CREB phosphorylation within the nuclei was completely prevented (Figures 6b and c). This demonstrates that cGKII regulates phospho-CREB clustering in the nucleus of retinal neurons. In addition, we injected lentiviruses coding for cGKII shRNAs in ovo and the resulting in vivo cGKII knockdown (Figure 6d) completely abrogated phospho-CREB downregulation (E6–E9; Figure 6e, upper panels) or upregulation (E8–E11; Figure 6e, bottom panels) induced by SNAP. Interestingly, NO effect in regulating phospho-CREB varied among retinal layers in E6–E9 and E8–E11. In E6–E9, SNAP regulated phospho-CREB staining via cGKII in the photoreceptor layer (ONL; Figure 6f) and in the inner nuclear layer (INL; Figure 6g) but not in the ganglion cell layer (GCL; Figure 6h). In E8–E11, NO/cGKII effect in increasing phospho-CREB was restricted to the INL (Figure 6j) and GCL (Figure 6k) with no effect in the ONL (Figure 6i).

Development-dependent retinal CREB activity is mediated by cGKII in vivo. Phospho-CREB and cGKII colocalize in retinal neurons. Cultured purified neurons (a.1) or retinal sections (a.2) were stained against CREB (green (E6) or cyan (E8)) and cGKII (red (E6) or magenta (E8)) and observed in a confocal microscope. Calibration bars=10 μm in a.1 and 50 μm in a.2. In b and c, NO decreases or increases nuclear CREB activity via cGKII. E6C3 and E8C3 (respectively) mixed retinal cultures were immunostained against pCREB Ser133 and processed for nuclear colocalization with DAPI. In d, cGKII is knocked down in vivo. Lentiviruses coding for cGKII shRNAs or the empty vector pLKO were injected in the air chamber of chicken eggs with E5 embryos and incubated up to E9–E11. Retinas were immunostained for cGKII. Knockdown51.5% in E5–E9 and 78.3% in E8–E11. Calibration bar=50 μm. In e–k, NO differentially regulates phospho-CREB in retinal layers via cGKII. Lentiviruses coding for cGKII shRNAs were injected (as in d), and then SNAP was administrated at E6 (upper panels) or at E8 (bottom panels). Eggs were incubated for further 3 days (E9 or E11, respectively) and pCREB immunohistochemistry was performed. Calibration bars=50 μm. ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer. In k, NO regulates in vivo retinal tissue apoptotic labeling via cGKII. cGKII shRNAs were injected (as above), and then SNAP was administrated at E6 (upper) or at E8 (bottom). Cleaved caspase-3 staining=red staining in E6–E9 and green staining in E8-E11. Calibration bars=50 μm. Data represent the mean±S.E.M. (error bars). *P<0.05, **P<0.01, ***P<0.001 in relation to pLKO in figures (b–l); #P<0.05, ##P<0.01, ###P<0.001 in relation to pLKO with SNAP in figures (b and c, e–g, j–l); N=3–4, one-way ANOVA
The extent of cleaved caspase-3 in conditions where cGKII knockdown was performed in the retina was also measured (Figure 6l). Remarkably, in E6–E9 retinas, SNAP increased cleaved caspase-3 labeling in control conditions (pLKO), while this effect was abrogated in retinas where cGKII was knocked down (Figure 6l, upper panels). On the other hand, in E8–E11 retinas, SNAP injections decreased cleaved caspase-3 immunolabeling significantly and cGKII knockdown prevented SNAP effect in decreasing cleaved caspase-3 staining (Figure 6l, bottom panels).
The protein kinase AKT is directly involved in neuronal survival.40 Moreover, it has been demonstrated that AKT may translocate to the nucleus and upregulate protective pathways.41, 42, 43 In different neuronal paradigms, AKT-dependent survival may rely on downstream CREB activity in the nucleus.44, 45 Furthermore, it has been reported that NO regulates AKT activation and nuclear translocation via cGKII in retinal neurons.22 We then decided to evaluate the role played by AKT in CREB phosphorylation regulated by cGKII in the nuclei. Knocking down cGKII with shRNAs was sufficient to increase nuclear phospho-AKT accumulation in E6 markedly (Figure 7a). Conversely, cGKII shRNAs reduced nuclear phospho-AKT levels in E8 significantly (Figure 7b). Interestingly, in ovo injections of SNAP reduced phospho-AKT nuclear staining in E6–E9 period, an effect abrogated by knocking down cGKII in vivo (Figure 7c, left panels). On the other hand, SNAP administration enhanced nuclear phospho-AKT levels in E8–E11 period and cGKII shRNAs also prevented this effect in vivo (Figure 7c, right panels). These data suggest that the NO/cGKII system regulates nuclear AKT activity in a development-regulated manner. Next, the impact of nuclear AKT on the regulation of cGKII-mediated CREB phosphorylation and neuronal survival was assessed in E6 or E8 neurons. Interestingly, cGKII shRNA robustly increased both nuclear phospho-CREB levels (Figure 7d) and cell survival (Figure 7f) when compared with control (pLKO) conditions in E6 cultures. Noteworthy, inhibiting AKT with triciribine completely blocked the effect of cGKII shRNA in E6 (Figures 7d and f). Besides, SNAP-mediated nuclear phospho-CREB increase (Figure 7e) or neuronal survival (Figure 7g) in E8 was also dependent on AKT activity since triciribine blocked both effects (Figures 7e and g). Collectively, these data may indicate that NO/cGKII reduce (E6) or enhance (E8) nuclear AKT activity to regulate CREB-dependent neuronal survival in the developing retina.

NO/cGKII regulate phospho-CREB via nuclear AKT activity. In a and b, cGKII regulates AKT nuclear accumulation in E6 and E8. Mixed cultures (a, E6; b, E8) were transduced with pLKO (CT; empty vector) or cGKII shRNA (sh cGKII). SNAP was administrated from C0 up to C2. AKT nuclear staining was evaluated in a confocal microscope. Calibration bar =10 μm. Data represent the mean±S.E.M. (error bars). *P<0.05; **P<0.01 in relation to pLKO in a and b; N=3, t-test. In c, NO/cGKII regulate nuclear AKT in vivo. Lentiviruses coding for pLKO (control) or cGKII shRNA were injected in ovo as in Figure 6d. SNAP was injected form e6 to E9 (left panels) or from E8 to E11 (right panels). The LAS AF software determined nuclear AKT staining. AKT nuclear intensity is shown in a rainbow color-coded map. Calibration bar=50 μm. Data represent the mean±S.E.M. (error bars). *P<0.05 in relation to pLKO untreated; #P<0.05 in relation to pLKO+SNAP; N=3, one-way ANOVA. In d and e, nuclear AKT regulates CREB activation. E6 (d) and E8 (e) mixed cultures were treated as above. Phospho-CREB immunostaining was evaluated in a confocal microscope. Tricib=triciribine (AKT inhibitor; 500 nM). Calibration bars in d and e=10 μm. Data represent the mean±S.E.M. (error bars). *P<0.05 in relation to pLKO in E6 or CT in E8; #P<0.05 versus cGKII shRNA; ##P<0.001 versus SNAP; N=3, one-way ANOVA. (f and g) AKT regulates NO-induced cell death or survival. Mixed E6 (f) or E8 (g) cultures were treated as above and cell death determined by EthD1 staining. Calibration bars=10 μm. In h–o, CREB1 regulates NO/cGKII-dependent neuronal death or survival via AKT. CREB1 was knocked down in E6 (h) or E8 (i) mixed cultures using shRNAs. Calibration bars in h and i= 10 μm. CREB1 knockdown enhances cell death in both E6 (j) and E8 (m) mixed cultures. In pLKO (control empty vector) cultures, NO promotes cell death in E6 (k) and enhances cell survival in E8 (n). In E6 cultures, knocked down for CREB1 (l) SNAP cannot increase cell death further while in E8 CREB1 shRNA completely prevents SNAP-mediated cell survival (o). Cell death was assessed by EthD1 labeling. Data represent the mean±S.E.M. (error bars). *P<0.05 in relation to pLKO (m) or CT (k and n); **P<0.01 in relation to pLKO; N=3, t-test. In p, hypothetical model describing the signaling pathways triggered by NO in E6 and E8 to regulate retinal cell viability during early retinal development
To further confirm the involvement of CREB in neuronal death or survival regulated by NO/cGKII, shRNAs were used to knockdown CREB1 expression in E6 or E8 retinal cultures (Figures 7h and i). Knocking down CREB1 in E6 or E8 was sufficient to increase cell death robustly (Figures 7j and m). Moreover, as cell death was already high in E6 cultures knocked down for CREB1, SNAP could no longer increase neuronal death in such conditions (Figures 7k and l). However, CREB1 shRNA completely abrogated SNAP-mediated neuronal survival in E8 cultures (Figures 7n and o). We therefore suggest that CREB1 is a downstream target of the NO pathway to modulate retinal cell viability in E8 and to promote a caspase-dependent apoptotic cell death in E6 retinal neurons.
Discussion
In this study, we present evidence that NO, derived either from SNAP or from the nNOS substrate L-arginine, acts through sGC and cGKII to promote retinal neuronal death at E6 and to increase neuronal survival at E8. NO activated the transcription factor CREB in E8 neurons, whereas in E6 neurons it downregulated CREB activity in a cGKII/AKT-dependent manner. E6 neurons displayed increased calcium influx, nNOS activation and produced higher amounts of NO as compared with E8 neurons. Therefore, these observations suggest that in E6 retinas, high NO levels repress the production of survival factors directly controlled by CREB1 (Figure 7p), whereas at E8, low/moderate NO increases CREB1-mediated transcription of neuroprotective genes, implying that in early retinal development, concatenated NO-cGKII-AKT-CREB1 signaling orchestrate a fine-tuned development of retinal neurons.
In E6, both cone photoreceptors and MNs were highly susceptible to NO given that low or high concentration of SNAP increased neuronal loss significantly. On the contrary, MNs were responsive to SNAP-induced cell survival in E8, whereas cone photoreceptors were not. Since in E6 cultures there is a high proportion of cone photoreceptors in relation to MNs,31 one could regard that only cone photoreceptors would be responsive to SNAP in E6, but this is not the case. Interestingly, in E8 cultures, MNs are more numerous than photoreceptors, and SNAP does not regulate the size of cone photoreceptor population but only MNs survival. These data suggest that cone photoreceptors are not exclusively susceptible to NO cytotoxic effects in E6 cultures, but in E8 NO exclusively protects MNs in culture. Furthermore, in vivo experiments suggested that NO-induced CREB downregulation or upregulation is specific for retinal layers in E6 or E8 intact tissue, demonstrating that NO may physiologically regulate developmental cell death or survival in the retina via a downstream regulation of CREB activity.
We showed that SNAP was highly effective in promoting cell death in E6 cultures and increasing cleaved caspase 3 staining in vivo. Our data suggest that NO acts primarily through the cGMP/sGC/cGKII system to promote retinal cell apoptosis. This fact is interesting since NO decreased cleaved caspase 3 staining and apoptosis in E8 retinas using this same signaling pathway. SNAP-induced neuronal cell loss is a long-term event, which is typical of apoptotic cell death. Based on cell survival data and the proliferation profile of E6 cultured cells,27 we ruled out unspecific effects of SNAP in the proliferation experiments. Indeed, apoptosis induced by NO appears not to be related to a previous cell cycle arrest of mitotically active neurons since, although SNAP reduced [3H]-thymidine incorporation, DTT prevented its effect but ODQ did not while ODQ, but not DTT, prevented SNAP-mediated cell loss in E6, indicating that NO modulates apoptosis and proliferation through distinct signaling mechanisms. In agreement, previous data suggest that the proliferation of retinal cells is under the control of signaling pathways modulated by S-nitrosylation.11
It was previously established that cGKI and cGKII are downstream targets of NO signaling in neuronal cells.16 In addition, cGKII activity has been shown to modulate the trafficking of AMPA receptors and LTP in a cGMP-dependent manner in hippocampal slices.20 Nonetheless, cGK activation has been shown to promote neuronal cell death in a mouse model of retinitis pigmentosa.46 We have shown that cGKII precisely controls neuronal survival as a downstream relay for NO in the retina. We have previously demonstrated that cGKs regulate the activation of Src in retinal neurons, a pathway associated with AMPA-induced nerve cell loss.15 Here, we have demonstrated that the downstream pathway activated by cGKII is critical in determining the fate of developing retinal neurons in both E6 and E8 retinas.
Pharmacological data showed that cGK-induced Src activation in the retina also couples to ERK phosphorylation.15 Moreover, ERKs are known to modulate the late phase of CREB activation in neuronal cells.47 We previously demonstrated that neuronal-derived NO increased phospho-CREB in cultured retinal cells in an ERK-dependent manner.23 On the other hand, nNOS activity has been associated with both glutamate-induced excitotoxicity in cortical neurons48 and AMPA-induced apoptosis in the retina.15 We showed that calcium influx, nNOS activity and NO production were high in E6 when compared with E8 retinas. Since nNOS is a calcium-dependent enzyme and high NO concentration may acutely promote neuronal damage,49 these data could explain why NO was neurotoxic in E6 but not in E8 retinas. Dose-response curves of SNAP in E6 and E8 were instructive in demonstrating that NO-dependent regulation of neuronal survival relies on NO amounts in both developmental periods. In such way, increasing NO from elevated to high concentration in E6 (using NO donors or L-Arg) strongly induced neuronal death. In E8, low/moderate NO is protective but high NO (using 150 or 300 μM SNAP) is also neurotoxic, corroborating the idea that the shift of NO action, from protective-to-toxic, is strictly dependent on its concentration in retinal neurons. Similarly, cytotoxic NO content in retinal neurons may also directly contribute to high cGMP accumulation and misbalanced cGKII activation, which leads to the extrusion of AKT from the nucleus and CREB downregulation. Conversely, it would be plausible to assume that low/moderate concentration of NO could promote CREB1 activation in E8 neurons via increased nuclear phospho-AKT accumulation, leading to neuroprotection, in a cGKII-dependent manner. Furthermore, cytotoxic doses of NO, in E6 or even in E8, were also capable of decreasing phospho-CREB in a cGK-dependent manner, indicating that a developmental switch in NO-dependent retinal cell survival may rely in the differential activation of cGKII by different concentrations of NO (high in E6 and low in E8).
CREB has a major role in neuronal survival.50 Particularly, CREB1 may be involved in neuronal survival and metabolism,51 including in photoreceptor degeneration.52 When Ser133 of CREB is phosphorylated, its transcriptional activity is turned on,53, 54 but phosphorylation of other residues also modulate CREB-dependent transcription in neurons.39 It was demonstrated that a decrease in CREB phosphorylation was linked to kainate-induced cell death in the mature retina55 and adenosine-induced cell death in the developing retina.27 Furthermore, FGF-induced CREB activation was proposed as a protective mechanism in a paradigm of light-induced photoreceptor degeneration28 and in NMDA-mediated retinal ganglion cell loss.56 This evidence supports our findings showing that in E6 retinal neurons, NO/cGKII decreased CREB phosphorylation and shRNA-mediated CREB1 downregulation was directly associated with the degeneration of retinal neurons. On the other hand, anti-apoptotic actions of NO/cGK in R28 retinal cell line was prevented by expressing a dominant-negative CREB protein57 and we showed here that NO may exert a neuroprotective effect through a cGKII-induced CREB1 upregulation in E8.
It has been suggested that CREB may function as a network hub acting through multiple events controlling Ser133 phosphorylation.25 Therefore, it would be reasonable to assume that CREB integrates contextual information from the developing retinal environment. As a direct consequence, pro-survival genes controlled by CREB1 could be upregulated by extracellular clues. An element that directly controls CREB activity and cell survival is the protein kinase AKT.44, 45 We showed here that NO/cGKII directly influenced nuclear AKT localization in E6 and E8 retinal neurons. Interestingly, NO/cGKII-dependent neuronal death or survival was directly associated with an AKT-regulated CREB activation in the nucleus. In E6 neurons, NO-activated cGKII promotes AKT extrusion from the nucleus and phospho-CREB decrease, whereas in E8, NO/cGKII promotes AKT accumulation within the nucleus to upregulate phospho-CREB. Accordingly, NO/cGKII could inhibit CREB1 to impair the transcription of neuroprotective genes in E6 retinal cells, whereas in the E8 period, NO/cGKII signaling would enhance CREB1 activation to directly promote cell survival (Figure 7p). Consequently, we consider that populations of retinal neuronal cells expressing both cGKII and CREB1 could be either positively or negatively selected by exposure to graded NO concentrations during embryonic retinal development.
To the best of our knowledge here we described the first evidence that NO/cGKII/AKT dualistically regulate survival events in the developing vertebrate retina via CREB1 activity. These data may impact on the current view of retinal neurodegenerative disorders with a cGMP/cGK-dependent component. Moreover, pharmacological or genetic targeting of the NO/cGKII pathway could be a feasible strategy to prevent cell loss in degenerative conditions of the embryonic or mature retina. Overall, this view deems that concatenated nNOS, sGC, cGKII, AKT and CREB1 activities have important physiological roles controlling the viability of neurons during retinal development.
Materials and Methods
Materials
Fertilized white Leghorn chicken eggs were obtained from a local hatchery and incubated at 38 °C in a humidified atmosphere up to the appropriate age. Penicillin, streptomycin, fetal bovine serum (FBS), trypsin, minimum essential medium (MEM), 199 medium, cPTIO (2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide, potassium salt), L-glutamine and TUNEL kits were purchased from Life Technology (São Paulo, Brazil). L-arginine, 8-Br cGMP (8-bromo cyclic GMP), YC-1 (3-(5¢-hydroxymethyl-2¢-furyl)-1-benzylindazole), zaprinast (1,4dihydro5 (2propoxyphenyl)-7H-1,2,3-triazolo-(4,5-d )pyrimidin-7-one) and KT5823 were from Calbiochem (Merck/Millipore, São Paulo, Brazil). SNAP, dimethyl sulphoxide, ODQ, L-NAME (NW-nitro-L-arginine methyl ester), glutaraldehyde 25%, DTT, poly-L-ornithine and shRNA plasmids prkg2 (TRCN0000194661 and TRCN0000195033) or creb1 (TRCN0000304386) were from Sigma (São Paulo, Brazil). [3H]-thymidine (5 Ci/mmol), PVDF membranes, anti-goat HRP-conjugated secondary antibody and ECL chemiluminescence kit were from GE Healthcare (São Paulo, Brazil). OCT embedding medium was from Sakura (Tokyo, Japan).
Antibodies
Primary antibodies: mouse anti-CREB (#9197), mouse anti-phospho-CREB Ser133 (#9196), rabbit anti-phospho-CREB Ser133 (#9191) were from Cell Signaling (Danvers, MA, USA) and their specificity for chick CREB has been described previously23, 27; rabbit anti-cGKII (ab110124) was from Abcam (Cambridge, MA, USA), rabbit anti-phospho-AKT Ser473 (#9271) was from Cell Signaling and the specificity of both antibodies in the chick was previously determined22; rabbit anti-phospho-nNOS Ser1417 (07–544) was from Millipore (Merck/Millipore) and its specificity was previously determined.15 Antibody labeling in chick retinal cells was re-validated (Supplementary Figure 1). Rabbit anti-cleaved caspase 3 was a kind gift from Dr. Ana LM Ventura and its specificity in the chick retina was described elsewhere.58
Primary retinal cultures
Purified neuronal cultures and drug exposure were carried out as described previously.27 In brief, E6 or E8 chick retinas were dissected, suspended in calcium and magnesium-free Hank’s buffer balanced salt solution (HBSS) and trypsinized at 37 °C for 9 min. Retinal tissue was mechanically dissociated using a fire-polished Pasteur pipette, suspended in medium 199 (containing 2.5% (v/v) FBS, supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin and 2 mM glutamine) and seeded onto 40 mm or 12-well plastic culture dishes pre-treated for 2 h with poly-L-ornithine (50 μg/ml). Cells were diluted to 832 cells/mm2. Cultures were incubated at 37 °C in an atmosphere of 5% CO2/95% air. Cell composition and characterization in E6 and E8 purified cultures has been described previously.31 In E6 cultures, the cell composition was 25–30% neurons (amacrine-like cells) to 75–80% cone photoreceptors and in E8 20–30% cone photoreceptors to 70–80% amacrine like-cells.31 Counting of viable cells was performed as previously described.27 Cells in vitro for 2 h (C0) or 24 h (C1) were treated with different drugs at the indicated concentrations. Purified cultures were used in cell counting, AO/EB exclusion, thymidine incorporation and DAF-FM-DA experiments.
Mixed primary cultures of retinal cells were prepared as previously described.15, 23, 27 E6 or E8 chick embryo retinas were suspended in calcium and magnesium-free HBSS and digested with 0.1% trypsin (w/v) for 17 min at 37 °C. Cells were suspended in MEM (supplemented with 3% FBS (v/v), 100 U/ml penicillin, 100 μg/ml streptomycin and 2 mM glutamine), dissociated using a Pasteur pipette, and seeded onto 24- or 12-well culture plastic dishes in a density of 2 × 105 cells/mm2. Cells were maintained at 37 °C in a humidified incubator with 5% CO2, 95% air. Cellular composition and cell characterization in these cultures showed that 80–85% were neurons and 15–20% were glial cells with a Müller cell phenotype.14 Experiments with lentiviruses shRNAs in vitro were all carried out in mixed cultures since lentiviral infection used here was highly toxic to purified neurons for long incubation periods. For western blotting, EthD1 and MTT data mixed cells were also used.
Lentiviruses production and retinal culture infection
Low passage HEK293T cells were seeded in 90 mm culture dishes. When cultures reached ∼80% confluence cells were co-transfected overnight with viruses-producing plasmids using Lipofectamine2000 (Invitrogen, Life Technology) according to the manufacturer’s instructions. Transfection ratios were as follows: 6 μg of shRNA plasmids to 3 μg of psPAX2 to 3 μg of VSVG (2 : 1 : 1). The next day, normal growth media replaced transfection media and cells were cultivated for an additional 48 h. Next, media with viral particles were collected, centrifuged at 1500 r.p.m. for 5 min, and the supernatant was collected into new tubes. Cultured retinal cells were infected with viral supernatants at C0 (2 h after culturing) overnight. At C1 (24 h after culturing), normal growth media replaced infection media and cells were cultivated up to C4.
Detection of retinal cell death with fluorescent dyes
E6 and E8 purified neuronal cultures were cultivated on coverslips pretreated overnight with poly-L-ornithine (50 μg/ml). At C4, cultures were treated with a solution of 1 : 2 AO (50 ng/ml) and 1 : 2 EB (50 ng/ml) directly in the culture medium for 30 s. Then, coverslips were quickly mounted and visualized under a Nikon inverted fluorescence microscope (Tokyo, Japan) at 460 and 580 nm wavelengths, with cultured cells still alive. Photomicrographs were obtained from 10 randomly chosen coverslip fields per experimental group. Cell death index was calculated by the fraction of cells with condensed nuclei, labeled with EB, divided by the total number of cells in each field, labeled with AO.
In the case of EthD1 labeling, cells were washed 2 × with HANK’s and incubated with a solution of EthD1 (1 : 2000) in HANK’s for 30 min. Then cells were fixed with 1% paraformaldehyde, coverslips were mounted in DAKO glycergel and visualized in a Leica TCS SP5 II confocal microscope (Leica Microsystems, Wetzlar, Germany). Cell viability was evaluated by counting the number of EthD1-positive cells in six different microscope fields per experimental group. Experiments were always run in duplicate.
MTT viability assay
E6 or E8 mixed cultures were incubated with MTT for 4 h at 37 °C and washed twice in Hank’s solution. Cultures were next incubated with SNAP (100 μM) for 96 h. The dye produced by viable cells was quickly dissolved in acid/alcohol solution (0.6% (v/v) chloridric acid in isopropanol) and the optical density was measured at 570 nm.
Calcium-45 influx in retinal neurons
Cultured retinal neurons (either E6C1 or E8C1) were incubated with 1 μCi [45Ca2+] radionuclide in HBSS for 2 min at 37 °C. Next, cells were quickly washed with HBSS and lysed with water. Radioactivity in cell lysates was determined in a liquid scintillation counter. CPM normalization was performed by an estimate of mean neuronal cells in each condition and was represented as CPM per cell number.
[3H]-thymidine incorporation was performed as previously described.27
Western blotting
For detection of the phosphorylation level of the indicated proteins, cultures were washed in HBBS, scraped off from culture dishes using 50–100 μl of RIPA buffer with protease inhibitor cocktail and the material was sonicated and protein content was determined by the BCA method. Samples were submitted to 9% SDS-PAGE, the proteins (45 μg/lane) were transferred to PVDF membranes, which were next incubated overnight with specific antibodies (rabbit anti-cGKII (1 : 500), rabbit anti-phospho-CREB Ser133 (1 : 2000), mouse anti-CREB (1 : 1000), rabbit anti-phospho-nNOS Ser1417 (1 : 500) and rabbit anti-beta-tubulin (1 : 100 000)). Subsequently, membranes were washed in TBS buffer (20 mM Tris; 200 mM NaCl), pH 7.6, incubated with peroxidase-conjugated secondary antibody and developed using an ECL chemiluminescence kit. Images were acquired in a ChemiDoc XRS+ System (Bio-Rad, Hercules, CA, USA), exported using Image Lab software (Bio-Rad) and quantified by FIJI software (National Institute of Health, Bethesda, MD, USA).
Immunocytochemistry and colocalization data
Coverslips with 1% (w/v) paraformaldehyde-fixed retinal neuronal cultures were washed 2 × 5 min in PBS and incubated for 1 h with blocking solution (5% BSA (w/v), 5% FBS (v/v) and 0.1% Triton X-100 (v/v) in PBS). Next, primary antibodies were added, and the coverslips were maintained in a humidified chamber for 2 h (rabbit or mouse anti-phospho-CREB Ser133 1 : 100; rabbit anti-cGKII 1 : 50). Afterward, coverslips were washed 3 × 10 min with PBS and incubated with secondary antibodies (anti-rabbit Alexa 568 or anti-mouse Alexa 488 and anti-mouse Alexa 594 all in 1 : 200 dilution) for 2 h. Next, coverslips were washed 3 × 10 min with PBS, incubated for 1 min with a solution of 1 mg/ml DAPI and rinsed twice in PBS. Then coverslips were mounted with DAKO glycergel and visualized in a Leica TCS SP5 II confocal microscope. Quantification of the fluorescence intensity in experimental groups was determined using the LAS AF software (Leica Microsystems). Images were acquired using a HCX Plan Apo 63x/1.4-0.6 NA oil immersion objective in 16-bit sequential mode using bidirectional TCS mode at 100 Hz and the pinhole was kept at 1 airy. The LAS AF software processed images using thresholded background (+30% offset for both channels) and thresholded foreground (+20–35% offset for DAPI channel and +12–28% offset for phospho-CREB or phospho-AKT channel). Values corresponding to phospho-CREB/DAPI or phospho-AKT/DAPI pixels colocalization puncta were developed by LAS AF software and statistically evaluated by the Prism software (La Jolla, CA, USA).
Intact retina experiments and NO quantification by DAF-FM-DA labeling
E6 or E8 embryos had their retinas dissected at 4 °C and equilibrated in HBSS for 20 min at 37 °C. Next, the intact retinas were incubated with DAF-FM-DA (5 μM) in HBSS for 1 h in the dark. The retinas were then washed 2 × with HBSS and fixed with 4% paraformaldehyde for 1 h. Afterward, retinas were flat-mounted and visualized under a Leica TCS SP5 II confocal microscope. DAF-FM-DA quantification was determined using the LAS AF software (Leica Microsystems). DAF assay in purified E6 or E8 cultures were performed as previously described.15
In vivo experiments and tissue preparation
Lentiviruses carrying cGKII shRNAs or the empty vector pLKO (control) were injected in eggs with 5-day-old chicken embryos at the eggs’ air chamber (150 μl of lentiviruses-containing medium per egg) and incubated for the appropriate time for experiments. Next, E6 or E8 chicken eggs were injected with 60 μl of SNAP (100 mM) in sterile water. The egg volume was approximately 60 ml and so the estimated concentration of SNAP in the retina was approximately 100 μM. Embryos injected at E6 and E8 were returned to the incubator until being killed 3 days later (at E9 or E11, respectively). Vehicle groups were obtained using eggs injected with 60 μl of sterile water. Immediately after killing, retinas were dissected in ice-cold CMF (for western blotting) or fixed in 4% paraformaldehyde (for immunohistochemistry).
Immunohistochemistry
Eyes cup were fixed by immersion in 4% paraformaldehyde in 0.16 M phosphate buffer (PB), pH 7.2, for 1 h. The tissue was rinsed with PB and then cryoprotected using a two-sucrose gradient in a row (15 and 30%). After 24 h, retinas were mounted in OCT embedding medium, frozen and cryosectioned. Sections perpendicular to the vitreal surface (12 μm) were collected on gelatinized slides. Sections of the tissue from control and from different treatment conditions were collected on the same glass slide. The slides were stored at −20 °C until processed for TUNEL, rabbit anti-phospho-CREB (1 : 100), mouse anti-CREB (1 : 100), rabbit anti-cGKII (1 : 100) and rabbit anti-cleaved caspase-3 (1 : 50) immunohistochemistry. Next, retinas were washed 3 × 10 min with PBS and incubated for 1 h with blocking solution (3% bovine serum albumin, 3% FBS and 1% Triton X-100 in PBS). Primary antibodies were incubated and the material maintained in a humidified chamber overnight. In the next day, retinal sections were washed 3 × 10 min with PBS and secondary antibodies (1 : 300 anti-rabbit Alexa568 or Alexa488 and 1 : 300 anti-mouse Alexa 594) were incubated for 2 h. After that, cells were washed 3 × 10 min with PBS, 1 × 10 min with TBS, incubated for 40 s with a solution of 1 : 1000 diamidino-2-phenylindole and rinsed twice in TBS. Slides were cover slipped using DAKO glycergel and visualized under a Leica TCS SP5 II confocal microscope. Images were processed and quantified using the LAS AF software (Leica Microsystems). In the in vivo data, phospho-CREB, and phospho-AKT intensity or cells positive for cleaved caspase 3 were corrected as a function of retinal area in μm2.
Statistical analysis
Data are represented as the mean±S.E.M. in all histograms. Statistical significance was evaluated in the non-normalized (%) data by unpaired Student’s t-test or one-way ANOVA followed by the Bonferroni post-test, using Graph Pad Prism 5.0 software (La Jolla, CA, USA) for Mac.
Abbreviations
- AMPA:
-
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- AKT:
-
V-Akt murine thymoma viral oncogene
- cGK:
-
protein kinase, CGMP-dependent
- CREB:
-
cyclic AMP-responsive element-binding protein
- DAF-FM-DA:
-
4-amino-5-methylamino-2',7'-difluorofluorescein diacetate
- DTT:
-
dithiothreitol
- ERK:
-
extracellular regulated kinase
- EthD1:
-
ethidium homodimer-1
- FGF:
-
fibroblast growth factor
- L-NAME:
-
Nω-nitro-L-arginine methyl ester hydrochloride
- NO:
-
nitric oxide
- nNOS:
-
neuronal nitric oxide synthase
- ODQ:
-
1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one
- sGC:
-
soluble guanylyl cyclase
- SNAP:
-
S-nitroso-N-acetyl-D,L-penicillamine
- Src:
-
Src avian sarcoma (Schmidt-Ruppin A-2) viral oncogene homolog
- TUNEL:
-
terminal deoxynucleotidyl transferase dUTP nick end labeling
References
Marquardt T, Gruss P . Generating neuronal diversity in the retina: one for nearly all. Trends Neurosci 2002; 25: 32.
Yang XJ . Roles of cell-extrinsic growth factors in vertebrate eye pattern formation and retinogenesis. Semin Cell Dev Biol 2004; 15: 91–103.
Wiesinger H . Arginine metabolism and the synthesis of nitric oxide in the nervous system. Prog Neurobiol 2001; 64: 365–391.
Knowles RG, Palacios M, Palmer RM, Moncada S . Formation of nitric oxide from L-arginine in the central nervous system: a transduction mechanism for stimulation of the soluble guanylate cyclase. Proc Natl Acad Sci USA 1989; 86: 5159–5162.
Krumenacker JS, Hanafy KA, Murad F . Regulation of nitric oxide and soluble guanylyl cyclase. Brain Res Bull 2004; 62: 505–515.
Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH . Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol 2001; 3: 193–197.
Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS . Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol 2005; 6: 150–166.
Liu L, Stamler JS . NO: an inhibitor of cell death. Cell Death Differ 1999; 6: 937–942.
Boehning D, Snyder SH . Novel neural modulators. Annu Rev Neurosci 2003; 26: 105–131.
Peunova N, Enikolopov G . Nitric oxide triggers a switch to growth arrest during differentiation of neuronal cells. Nature 1995; 375: 68–73.
Magalhães CR, Socodato RES, Paes-de-Carvalho R . Nitric oxide regulates the proliferation of chick embryo retina cells by a cyclic GMP-independent mechanism. Int J Dev Neurosci 2006; 24: 53–60.
Mejía-Garcia TA, Paes-de-Carvalho R . Nitric oxide regulates cell survival in purified cultures of avian retinal neurons: involvement of multiple transduction pathways. J Neurochem 2007; 100: 382–394.
Cossenza M, Paes-de-Carvalho R . L-arginine uptake and release by cultured avian retinal cells: differential cellular localization in relation to nitric oxide synthase. J Neurochem 2000; 74: 1885–1894.
Portugal CC, da Encarnação TG, Socodato R, Moreira SR, Brudzewsky D, Ambrósio AF et al. Nitric oxide modulates sodium vitamin C transporter 2 (SVCT-2) protein expression via protein kinase G (PKG) and nuclear factor-kappaB (NF-kB). J Biol Chem 2012; 287: 3860–3872.
Socodato R, Santiago FN, Portugal CC, Domingues AF, Santiago AR, Relvas JB et al. Calcium-permeable α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors trigger neuronal nitric-oxide synthase activation to promote nerve cell death in an Src kinase-dependent fashion. J Biol Chem 2012; 287: 38680–38694.
Hofmann F, Feil R, Kleppisch T, Schlossmann J . Function of cGMP-dependent protein kinases as revealed by gene deletion. Physiol Rev 2006; 86: 1–23.
Feil R, Lohmann SM, de Jonge H, Walter U, Hofmann F . Cyclic GMP-dependent protein kinases and the cardiovascular system insights from genetically modified mice. Circ Res 2003; 93: 907–916.
Surks HK, Mochizuki N, Kasai Y, Georgescu SP, Tang KM, Ito M et al. Regulation of myosin phosphatase by a specific interaction with cGMP-dependent protein kinase IŒ±. Science 1999; 286: 1583–1587.
Arancio O, Antonova I, Gambaryan S, Lohmann SM, Wood JS, Lawrence DS et al. Presynaptic role of cGMP-dependent protein kinase during long-lasting potentiation. J Neurosci 2001; 21: 143–149.
Serulle Y, Zhang S, Ninan I, Puzzo D, McCarthy M, Khatri L et al. A GluR1-cGKII interaction regulates AMPA receptor trafficking. Neuron 2007; 56: 670–688.
Wang H-G, Lu F-M, Jin I, Udo H, Kandel ER, de Vente J et al. Presynaptic and postsynaptic roles of NO, cGK, and RhoA in long-lasting potentiation and aggregation of synaptic proteins. Neuron 2005; 45: 389–403.
Mejía-García TA, Portugal CC, Encarnação TG, Prado MA, Paes-de-Carvalho R . Nitric oxide regulates AKT phosphorylation and nuclear translocation in cultured retinal cells. Cell Signal 2013; 25: 2424–2439.
Socodato RES, Magalhães CR, Paes-de-Carvalho R . Glutamate and nitric oxide modulate ERK and CREB phosphorylation in the avian retina: evidence for direct signaling from neurons to Müller glial cells. J Neurochem 2009; 108: 417–429.
Lonze BE, Ginty DD . Function and regulation of CREB family transcription factors in the nervous system. Neuron 2002; 35: 605–623.
Shaywitz AJ, Greenberg ME . CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem 1999; 68: 821–861.
West AE, Griffith EC, Greenberg ME . Regulation of transcription factors by neuronal activity. Nat Rev Neurosci 2002; 3: 921–931.
Socodato R, Brito R, Calaza KC, Paes-de-Carvalho R . Developmental regulation of neuronal survival by adenosine in the in vitro and in vivo avian retina depends on a shift of signaling pathways leading to CREB phosphorylation or dephosphorylation. J Neurochem 2011; 116: 227–239.
O'Driscoll C, O'Connor J, O'Brien CJ, Cotter TG . Basic fibroblast growth factor-induced protection from light damage in the mouse retina in vivo. J Neurochem 2008; 105: 524–536.
Riccio A, Alvania RS, Lonze BE, Ramanan N, Kim T, Huang Y et al. A nitric oxide signaling pathway controls CREB-mediated gene expression in neurons. Mol Cell 2006; 21: 283–294.
Paes-de-Carvalho R, Maia GA, Ferreira JM . Adenosine regulates the survival of avian retinal neurons and photoreceptors in culture. Neurochem Res 2003; 2003: 1583–1590.
Adler R, Hatlee M . Plasticity and differentiation of embryonic retinal cells after terminal mitosis. Science 1989; 243: 391–393.
Bredt DS, Hwang PM, Snyder SH . Localization of nitric oxide synthase indicating a neural role for nitric oxide. Nature 1990; 347: 768.
Bredt DS, Snyder SH . Nitric oxide mediates glutamate-linked enhancement of cGMP levels in the cerebellum. Proc Natl Acad Sci USA 1989; 86: 9030–9033.
Arnelle DR, Stamler JS . NO+, NO., and NO− donation by S-nitrosothiols: implications for regulation of physiological functions by S-nitrosylation and acceleration of disulfide formation. Arch Biochem Biophys 1995; 318: 279–285.
Zeller A, Wenzl MV, Beretta M, Stessel H, Russwurm M, Koesling D et al. Mechanisms underlying activation of soluble guanylate cyclase by the nitroxyl donor Angeli's salt. Mol Pharmacol 2009; 76: 1115–1122.
Irvine JC, Ritchie RH, Favaloro JL, Andrews KL, Widdop RE, Kemp-Harper BK . Nitroxyl (HNO): the Cinderella of the nitric oxide story. Trends Pharmacol Sci 2008; 29: 601–608.
Hardingham GE, Arnold FJ, Bading H . Nuclear calcium signaling controls CREB-mediated gene expression triggered by synaptic activity. Nat Neurosci 2001; 4: 261–267.
Hardingham GE, Fukunaga Y, Bading H . Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci 2002; 5: 405–414.
Kornhauser JM, Cowan CW, Shaywitz AJ, Dolmetsch RE, Griffith EC, Hu LS et al. CREB transcriptional activity in neurons is regulated by multiple, calcium-specific phosphorylation events. Neuron 2002; 34: 221–233.
Datta SR, Brunet A, Greenberg ME . Cellular survival: a play in three Akts. Genes Dev 1999; 13: 2905–2927.
Lee SB, Xuan Nguyen TL, Choi JW, Lee KH, Cho SW, Liu Z et al. Nuclear Akt interacts with B23/NPM and protects it from proteolytic cleavage, enhancing cell survival. Proc Natl Acad Sci USA 2008; 105: 16584–16589.
Ahn JY, Liu X, Liu Z, Pereira L, Cheng D, Peng J et al. Nuclear Akt associates with PKC-phosphorylated Ebp1, preventing DNA fragmentation by inhibition of caspase-activated DNase. EMBO J 2006; 25: 2083–2095.
Hu Y, Yao J, Liu Z, Liu X, Fu H, Ye K . Akt phosphorylates acinus and inhibits its proteolytic cleavage, preventing chromatin condensation. EMBO J 2005; 24: 3543–3554.
Jain V, Baitharu I, Prasad D, Ilavazhagan G . Enriched environment prevents hypobaric hypoxia induced memory impairment and neurodegeneration: role of BDNF/PI3K/GSK3beta pathway coupled with CREB activation. PLoS One 2013; 8: e62235.
Simao F, Matte A, Pagnussat AS, Netto CA, Salbego CG . Resveratrol prevents CA1 neurons against ischemic injury by parallel modulation of both GSK-3beta and CREB through PI3-K/Akt pathways. Eur J Neurosci 2012; 36: 2899–2905.
Paquet-Durand F, Hauck SM, Van Veen T, Ueffing M, Ekström P . PKG activity causes photoreceptor cell death in two retinitis pigmentosa models. J Neurochem 2008; 108: 796–810.
Wu GY, Deisseroth K, Tsien RW . Activity-dependent CREB phosphorylation: convergence of a fast, sensitive calmodulin kinase pathway and a slow, less sensitive mitogen-activated protein kinase pathway. Proc Natl Acad Sci USA 2001; 98: 2808–2813.
Rameau GA, Tukey DS, Garcin-Hosfield ED, Titcombe RF, Misra C, Khatri L et al. Biphasic coupling of neuronal nitric oxide synthase phosphorylation to the NMDA receptor regulates AMPA receptor trafficking and neuronal cell death. J Neurosci 2007; 27: 3445–3455.
Brown GC . Nitric oxide and neuronal death. Nitric Oxide Biol Chem 2010; 23: 153–165.
Walton MR, Dragunow I . Is CREB a key to neuronal survival? Trends Neurosci 2000; 23: 48–53.
Fusco S, Ripoli C, Podda MV, Ranieri SC, Leone L, Toietta G et al. A role for neuronal cAMP responsive-element binding (CREB)-1 in brain responses to calorie restriction. Proc Natl Acad Sci USA 2012; 109: 621–626.
Beltran WA, Allore HG, Johnson E, Towle V, Tao W, Acland GM et al. CREB1/ATF1 activation in photoreceptor degeneration and protection. Invest Ophthalmol Vis Sci 2009; 50: 5355–5363.
Yamamoto KK, Gonzalez GA, Biggs WH 3rd, Montminy MR . Phosphorylation-induced binding and transcriptional efficacy of nuclear factor CREB. Nature 1988; 334: 494–498.
Gonzalez GA, Montminy MR . Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell 1989; 59: 675–680.
Mali RS, Zhang XM, Chintala SK . A decrease in phosphorylation of cAMP-response element-binding protein (CREBP) promotes retinal degeneration. Exp Eye Res 2011; 92: 528–536.
Takeda H, Kitaoka Y, Hayashi Y, Kumai T, Munemasa Y, Fujino H et al. Calcium/calmodulin-dependent protein kinase II regulates the phosphorylation of CREB in NMDA-induced retinal neurotoxicity. Brain Res 2007; 1184: 306–315.
Nagai-Kusuhara A, Nakamura M, Mukuno H, Kanamori A, Negi A, Seigel GM . cAMP-responsive element binding protein mediates a cGMP/protein kinase G-dependent anti-apoptotic signal induced by nitric oxide in retinal neuro-glial progenitor cells. Exp Eye Res 2007; 84: 152–162.
Anccasi RM, Ornelas IM, Cossenza M, Persechini PM, Ventura ALM ATP . induces the death of developing avian retinal neurons in culture via activation of P2X7 and glutamate receptors. Purinergic Signal 2013; 9: 15–29.
Acknowledgements
We thank Luzeli R de Assis and Sarah A Rodrigues for technical assistance and Edward B Ziff for critically reviewing the manuscript. This work was supported by grants from CNPq, CAPES, FAPERJ and PRONEX/FAPERJ. RS, RB, CCP and NAO were recipients of student fellowships from CAPES. RPC and KCC were research fellows from CNPq.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by G Melino
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article
Cite this article
Socodato, R., Brito, R., Portugal, C. et al. The nitric oxide-cGKII system relays death and survival signals during embryonic retinal development via AKT-induced CREB1 activation. Cell Death Differ 21, 915–928 (2014). https://doi.org/10.1038/cdd.2014.11
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2014.11

{kind=link}