
Abstract
Control of apoptotic signalling pathways depends on the balance between proapoptotic and prosurvival molecules. The ‘inhibitor of apoptosis’ (IAP) proteins are negative regulators of apoptosis that function by inhibiting the executioners of cell death (caspases), or by blocking the pathways that activate them. The IAP proteins function as ubiquitin E3 ligases and possess protein–protein interaction domains. IAPs can promote the addition of ubiquitin to themselves and to the substrate proteins that they interact with either directly or indirectly through adaptor proteins. The balance between substrate and autoubiquitylation seems to be important for their activity. In this review, we describe the structural features of IAP proteins as they are currently understood, and discuss how each domain contributes to IAP function. It is clear that to advance our understanding of these complex proteins, we must decipher how the domains operate in concert and how each domain impacts on the activity of the other.
Similar content being viewed by others

Main
In mammals, two largely independent responses account for the activation of apoptosis. The ‘extrinsic’ pathway is initiated when specific ligands bind to cell-surface receptors of the tumour necrosis factor receptor (TNFR) family.1 The ‘intrinsic’ pathway is triggered by the disruption of cellular integrity and relies on the release of proapoptotic factors from the mitochondria.2 Initiation of either pathway ultimately leads to the activation of caspases – cysteine aspartic acid specific proteases that bring about destruction of the cell.3 The intrinsic pathway primarily depends on activation of caspase-9, whereas activation of caspase-8 is important for signalling from the TNFR protein family. On activation of caspase-8 or -9, downstream caspases, or ‘effector’ caspases, such as caspase-3 and -7 are cleaved and cells become committed to death because of proteolysis of key proteins. To prevent inappropriate death, cells possess a multitude of specific inhibitors that can restrain and/or block caspase activation. One such family is the ‘inhibitor of apoptosis’ (IAP) proteins, which was first identified in baculoviruses,4 and then found to be widespread with homologues in yeast, flies and mammals.5
To understand the mechanism by which IAP proteins inhibit apoptosis, attention was initially focused on the baculoviral IAP repeat (BIR) domains. These studies showed that BIR domains are multifaceted protein–protein interaction domains that can bind many proteins and are able to directly inhibit caspases.6, 7 Subsequently, the RING (really interesting new gene 1) domain was shown to be important, and like many proteins that contain RING domains, IAPs function as E3 ligases and promote the transfer of ubiquitin from E2 ubiquitin-conjugating enzymes to substrate proteins.7, 8 In addition, Yang et al.9 have shown that cIAP1 and XIAP can promote the transfer of ubiquitin to themselves, and that this activity relies on an intact RING domain.
Inhibitor of apoptosis proteins that regulate apoptosis have both BIR and RING domains, and are the focus of this review. Recent studies using novel therapeutic agents suggest that the BIR and RING domains are intimately linked, and that the E3 ligase activity of the RING domain is modulated in unexpected ways by the BIR domains.10, 11 Further studies have shown that IAPs also possess a ubiquitin-binding domain that binds to ubiquitin chains, and is important for their activity.12, 13 These studies point to an interplay between substrate recruitment and ubiquitylation, and suggest that cross-talk between the domains is more important than initially thought. Here, we review the structures of the individual domains and how these underpin our current understanding of IAP function.
Structures of the Building Blocks
BIR domain
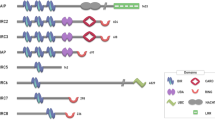
All IAP proteins have at least one BIR domain (Figure 1a). The BIR domain is ∼80 amino acids in length and is defined by conserved histidine and cysteine residues that coordinate a zinc ion.14, 15, 16, 17, 18, 19 The BIR domain comprises three short β-strands and four α-helices, and folds into a compact structure that includes a coordinated zinc ion (Figure 1b). Many IAPs have multiple BIR domains; the mammalian IAPs XIAP, cIAP1 and cIAP2 each contain three such domains at their N-terminus, whereas DIAP1 from Drosophila contains two BIR domains that are functionally equivalent to BIR2 and BIR3 of XIAP (Figure 1a).7 In addition to the core secondary structural elements, the most C-terminal BIR domains contain a further α-helix (α5) at their C-terminus (Figure 1b). Overall, the BIR domain sequences are strongly conserved from viral through to mammalian IAPs, and structurally, they are remarkable in their similarity (Figure 1c). For instance, structures of the three BIR domains from XIAP and two BIR domains from DIAP1 are available, and these overlay with a root mean square deviation (r.m.s.d.) of 1.0 Å over 72 residues (Figure 1c).6, 17, 18, 19, 20, 21 However, specific sequence differences underlie the distinct binding properties of each BIR domain (Figure 2), and the presence of multiple BIR domains confers, on IAPs, the ability to interact with a number of proteins (Figure 3a). These interactions serve to localise IAPs, and to either directly inhibit substrate proteins or recruit them for ubiquitylation, as discussed below.

Domain organisation of IAP proteins. (a) Schematic representation, drawn to scale, of several of the IAPs discussed in this review. (b) Structure of BIR3 from XIAP (PDB code 1g3f), coloured from N-terminus (blue) to C-terminus (red). (c) Superposition of the five unique BIR domains from XIAP and DIAP1 (PDB codes 2poi, 1i3o, 3cm2, 1se0 and 1jd5, respectively). Chains are coloured from N-terminus (blue) to C-terminus (red), and BIRs with unique C-terminal arrangements are labelled. (d) Structure of the cIAP2 RING domain homodimer (PDB code 3eb5)

The BIR domain, a rigid but versatile recruitment module. (a) Structure-based alignment of various BIR domains discussed in this review with interface residues and elements of secondary structure indicated. (b–e) The structures of BIR domains from XIAP (blue ribbon) bound to various binding partners, shown as surface representations. The BIR domain is shown in the same orientation and scale in all cases. (b) BIR2 bound to caspase-3 (PDB code 1i3o). (c) BIR3 bound to caspase-9 (PDB code 1nw9). (d) BIR3 bound to the four N-terminal residues (AVPI) of Smac/DIABLO (PDB code 1g73). The remainder of Smac/Diablo is omitted for clarity. The view on the left is oriented so that the BIR domain is positioned as for (b and c). The view on the right is rotated 90 degrees to display the fit of the IBM in the IBM-binding groove of BIR3 (right). (e) BIR1 bound to TAB1 (PDB code 2pop)

IAPs and their binding partners (a) Schematic representation displaying selected binding partners of the IAP proteins. *TAB-1 only binds to the BIR1 domain of XIAP. **TRAFs only bind to the BIR1 domain of cIAP1 or cIAP2. (b) The balance between substrate ubiquitylation and autoubiquitylation by cIAP1 and cIAP2 seems to be important. In unstimulated cells, cIAPs mediate ubiquitylation of RIP1 and NIK, leading to cell survival. Addition of IBM antagonist compounds enhances autoubiquitylation of cIAPs, resulting in their degradation, and thus, inhibiting the modification of RIP1 and NIK
A number of structures of BIR domains have been solved, either alone or in complex with binding partners. However, because only isolated BIR domains have been studied it is uncertain if BIR domains within an IAP polypeptide interact, or behave as ‘beads on a string’. Nonetheless crystal structures of individual BIR domains suggest that the BIR domains can associate. Notably, BIR1 from XIAP forms homodimers that depend on interaction of β3 and α3.20 In addition, BIR3 from XIAP forms dimers that depend on α5, although the interface is small (∼500 Å2) and a disulphide bridge appears to stabilise dimer formation.22 These studies suggest that BIR domains can associate but further studies are required to establish the relevance of these interactions, and to determine if contiguous BIR domains within a polypeptide interact.
RING domain
All IAPs that modulate the apoptotic pathways have a RING domain at their C-terminus, and this is required for dimerisation and for ubiquitin E3 ligase activity.23 The RING domain is a small (40 amino acid) domain that is defined by eight cysteine and histidine residues that coordinate two zinc ions (Figure 1d). The core structure of all RING domains is highly conserved, however, the surrounding residues vary and allow subclasses to be identified. In the case of IAPs the RING domain is at the C-terminus and is followed by exactly 13 residues. Mutagenesis studies have suggested that these residues are important for dimerisation.23 Equivalent residues are also required by other RING-containing proteins, such as mouse double minute proteins (MDMs), E3 ligases that determine the cellular levels of the tumour suppressor, p53.24 Structures of the RING domains from IAPs and MDMs have shown that the C-terminal residues have an extended conformation that includes β3, and interact across the dimer interface to mediate the dimer formation (Figure 1d).25, 26, 27 Both IAP heterodimers and homodimers have been reported, and although only the cIAP2 homodimer has been characterised in detail, it is likely that all pairings of IAP RING domains will adopt a similar structure.27
UBA domains
Following the final BIR domain in some IAPs is a short region that has recently been shown to have homology with ubiquitin-associated (UBA) domains that bind ubiquitin.12, 13 Although structures of the UBA domain from IAPs have not been determined, it is likely that they will also form a compact alpha helical bundle, similar to that of other UBA domains (for example, PDB codes 1vek and 2cpw). Already, NMR and mutagenesis experiments have shown that the hydrophobic conserved MGF/Y motif is important for binding ubiquitin, and that these residues interact with a conserved hydrophobic patch on ubiquitin.12, 13
CARD domains
A few IAPs also possess additional domains, such as the caspase recruitment domain (CARD), found in cIAP1 and cIAP2. The structure and function of the CARD in these IAPs has not been determined. However, given the conserved nature of this domain, it is highly likely that in IAPs the CARD forms a six-helix bundle, which serves as a protein–protein interaction motif.28
IAP Proteins – Direct Regulators of Caspase Activity
To understand how IAPs inhibit caspases, initial studies focused on the BIR domains of XIAP, as they directly inhibit both initiator and effector caspases.29 The molecular basis of effector caspase inhibition was revealed by crystal structures of the second BIR domain from XIAP (XBIR2), including an extended N-terminal linker, bound to caspase-730, 31 and caspase-3.32 In these structures, the residues N-terminal to XBIR2 extend into the substrate-binding groove of the caspase, blocking substrate entry in both caspase-3 and -7 (Figure 2a and b). Although the BIR domain is not visible in the caspase-7 complexes, the N-terminal peptide alone does not effectively inhibit caspases, suggesting that the BIR domain contributes to binding. Consistent with this, there are specific contacts between XBIR2 and caspase-3.32
Remarkably, inhibition of initiator caspases occurs in a distinct manner that requires the third BIR domain (XBIR3) of XIAP. Mutational analysis identified two regions that are required for inhibition, and showed that only processed caspase-9 is inhibited.33 Structural studies subsequently revealed two complimentary interactions between XBIR3 and caspase-9.21, 34 First, a tetrapeptide motif at the N-terminus of the small subunit of caspase-9 binds to a conserved surface groove of BIR3, and second, residues in α4 of BIR3 bind to the surface of caspase-9 that is required for dimerisation (Figure 2c).21 Fully active caspase-9 forms a homodimer, therefore, the two interactions with the BIR domain serve to hold caspase-9 in an inactive monomeric state.6
In contrast to XIAP, cIAP1 and cIAP2 are able to bind, but do not directly inhibit, the caspases.35 Instead, some have suggested that ubiquitylation of caspases by IAPs might be important,8 but the significance of this has not been established in mammals. However, recent studies in Drosophila show an important link.36 Using DIAP1, a Drosophila IAP protein that binds caspases through its BIR domains and flanking sequences, Ditzel et al.36 have shown that the abundance of active effector caspases is, in part, controlled by ubiquitylation. In its unmodified form DIAP1 ubiquitylates caspases poorly. However, after cleavage by the effector caspase drICE, DIAP1 is able to bind UBR domain-containing E3 ligases, such as UBR1, and polyubiquitylate the effector caspases DCP-1 and drICE. Although a proportion of this polyubiquitylation is K48-linked, drICE is not targeted for degradation, but rather has reduced catalytic potential. Thus, after activation of DIAP1 by caspases, DIAP1 in turn inactivates caspases – an elegant autoregulatory loop.
In mammals, on receipt of an apoptotic stimulus, restraint by the IAPs is relieved when the mitochondrial membrane is disrupted and proapoptotic proteins, such as Smac/DIABLO and HtrA2/Omi, are released.37, 38 These proteins have a conserved N-terminal sequence of four residues (AVPI in the case of Smac/DIABLO) that constitutes the IAP-binding motif (IBM). The IBM binds to a conserved groove on the BIR2 and BIR3 domains (Figure 2a and d).16, 17 This is the same groove that binds to the N-terminus of caspase-9, thus, when the IBM interacts with BIR3 it directly displaces caspase-9 thereby leading to its activation. In contrast, interaction of IBM-containing proteins with BIR2 sterically block effector caspases from binding to the BIR2-linker region.21, 34 The BIR domains of DIAP1 also have an IBM-binding pocket, and structural studies have shown that the IBM-binding site is conserved.29 Comparison of structures of BIR domains in the presence and absence of an IBM peptide show that the pocket is preformed, although two loops become ordered on binding,16 and in BIR2 from DIAP1 an additional helix (α6) forms on IBM peptide binding.18
Many tumors have elevated levels of IAPs and this correlates with a poor prognosis,39 suggesting that IAP antagonists may provide a therapeutic strategy for targeting these tumors. Importantly, it was shown using RNA interference methods that suppression of IAP protein levels, either directly stimulated cell death or increased the sensitivity of cells to undergo apoptosis induced by other therapeutic molecules.40 Given that the IBM tetrapeptide could promote caspase activity and sensitise cancer cell lines to undergo apoptosis,41, 42 these studies suggested that mimicking the function of Smac/DIABLO had therapeutic potential.43, 44 Several groups developed small molecule peptidomimetics of the IBM that bind selectively and with high affinity to the BIR domains of IAPs,43 and structures of compounds bound to BIR domains showed that they contacted the same residues in the IBM-binding pocket.45 Several of these compounds have been shown to stimulate cell death in various tumour cell lines as well as promoting tumour regression in model systems, and it was presumed that XIAP was the primary target of these compounds.43, 44
IAP Proteins – Modulators of Signalling Pathways
In addition to their roles as direct caspase regulators, mammalian IAPs have been shown to interact with adaptor proteins, and as a consequence, they are associated with several important signalling pathways. The first BIR domain is essential. In the case of XIAP, BIR1 binds to TAB1, an upstream component required for the activation of the TGF-β-activating kinase complex.20 This study not only showed that residues in α2 and α3 of XBIR1 are important for interaction with TAB1 (Figure 2a and e), but also that BIR1 forms a homodimer and this is important for activity. Other studies have shown that the BIR1 domain of cIAP1 and cIAP2 is important for their recruitment to the TNF receptor complexes.46, 47 The BIR1 domain of cIAP1 and cIAP2 interacts with TNF receptor-associated factors (TRAFs), adaptor proteins that are associated with TNFR complexes.48 Structures of these complexes have not been determined and the interaction site in TRAF2 is unknown, but mutagenesis has identified residues in α1 of BIR1 that are important for binding to TRAF2 (Figure 2a). These residues differ in BIR1 of XIAP, and this probably accounts for the selective binding of TRAFs to cIAPs.46, 47
Early studies suggested that cIAP1 mediated the ubiquitylation of TRAF2 in response to TNF binding and that this was important for signalling,49 but the precise role of cIAPs was uncertain. Recent studies using the IBM mimetic compounds have refocused the attention on the role of cIAPs, because their action is independent of caspase-9 and instead depends on caspase-8. This indicates that the antagonists trigger apoptosis mediated by the extrinsic apoptotic pathway.10, 11, 50 Rapid degradation of cIAP1 and cIAP2 by the proteasome seems to be critical to the response, and surprisingly loss of cIAPs ultimately leads to NF-κB activation and TNFα secretion. Caspase-8 activation then occurs in response to TNFα signalling and cell death ensues (Figure 3b).10, 11 Mutation of the RING domain in cIAP1 and cIAP2 abrogates E3 ligase activity and blocks the degradation of cIAP1 in response to antagonists, as does mutation of the IBM-binding groove. This points to an essential role for the RING domain in antagonist responses.10
How does loss of the cIAPs promote apoptosis? Signalling by the members of the TNFR superfamily depends on the recruitment of specific intracellular adaptor proteins that activate distinct signal transduction pathways. IAPs appear to modulate the composition of the TNFR signalling complex, and therefore, they can determine the cellular response to ligand binding.51, 52, 53 For example, cIAPs can modify the activity of receptor-interacting protein 1 (RIP1). The presence of K63-linked ubiquitin chains on RIP1 promotes the assembly of prosurvival complexes, and cIAPs are responsible for the attachment of these chains (Figure 3b).51, 54 Loss of cIAPs, as occurs on addition of IBM mimetics, means that RIP1 is no longer K63-ubiquitylated and no longer associates with the prosurvival kinase, TAK1. RIP1 instead interacts with caspase-8, leading to cell death.51 cIAP abundance also seems to be important for determining the levels of NF-κB-inducing kinase (NIK), another component that is required for TNFR superfamily signalling.10, 11 In resting cells, a TRAF2–TRAF3 complex brings NIK in close proximity to cIAP1 or cIAP2, promoting formation of K48-linked ubiquitin chains on NIK (Figure 3b), which leads to its degradation by the proteasome.52, 53 On receptor stimulation, TRAF2 mediates K63-linked ubiquitylation of cIAPs, which induces a change in cIAP substrate specificity. The cIAPs now promote K48-linked ubiquitylation of TRAF3 and TRAF2, causing their degradation, and thus, NIK is stabilised because it is no longer targeted by cIAPs.53 Therefore, ubiquitylation of IAPs seems to regulate their substrate specificity.
Together, these studies suggest that IAP-mediated ubiquitylation is critical, but a detailed understanding of the complexes formed by IAPs and the factors that determine whether K63- or K48-linked chains are formed, remain uncertain. The presence of the UBA domain that can interact with K48- or K63-linked ubiquitin chains adds another level of complexity that potentially influences the oncogenic potential of certain IAP proteins.13 Analysis of multiprotein receptor complexes is likely to be required to understand how both substrate recruitment and E3 ligase activity is controlled.
Regulating IAP RING E3 Ligase Activity
To characterise the molecular mechanism of ubiquitylation by IAP proteins studies have focused on the isolated IAP RING domains, which form dimers that depend on the integrity of the C-terminal residues (Figure 1d).23, 27 These residues are also important for the function of MDM2. Notably, although the RING domain of MDM2 with a mutated C-terminus is inactive and can no longer ubiquitylate itself or its substrate p53, activity is restored when MDM2 forms heterodimers with wild-type MDM4, even though MDM4 alone is not an active E3 ligase.24, 26 Thus, the C-terminus of MDM4 is able to restore the activity of mutant MDM2, showing that either directly or indirectly both monomers contribute to E3 ligase activity.
E2 recruitment is required for RING E3 ligase activity and IAPs have been reported to interact with a number of E2s.7 Unlike MDM2, which binds E2 enzymes with modest affinity,25 IAPs bind UbcH5b with at least μM affinity and the crystal structure of the isolated cIAP2 RING domain dimer bound to UbcH5b shows that both RING domains of the homodimer interact with an E2 molecule.27 It is uncertain whether both E2 : RING pairs are capable of promoting ubiquitin transfer, but comparison with other RING domains suggests that both E2-binding sites will not necessarily be occupied simultaneously (Figure 4a).55 Conserved hydrophobic residues that are required by many RING domains for activity and E2 recruitment are at the centre of the cIAP2 : RING interface. Surrounding residues also contribute to E2 recruitment, but these are less highly conserved, and are likely to contribute to selective binding.27 Although the subset of E2 enzymes that functionally interact with the IAP RING domains in vivo has not been clearly defined, studies with BRCA1 suggest that their identification may be important.

Snapshots of E3 ligase complexes (a) Model based on the structure of UbcH5b (grey) bound to the cIAP2 RING domain (green) (PDB code 3eb6). A ubiquitin molecule (red) has been modelled attached to cysteine 85 of UbcH5b to illustrate a charged E3:E2 unit before substrate ubiquitylation. (b) Regulation of RING E3 ligase activity by distant domains of CHIP. The two structures solved were (i) the U-box (yellow) bound to E2 (grey) (PDB code 2c2v), and (ii) the full CHIP protein, including U-box and TPR domains (purple) (PDB code 2c2l). Here, the two structures are overlaid to emphasise that in full-length CHIP the TPR domain of one monomer in the dimer blocks its own E2-binding site, allowing only one E2 molecule to bind the dimer as shown
Christensen et al.56 identified the E2 enzymes capable of binding BRCA1, and showed that of those able to bind and autoubiquitylate, a subset were only able to attach a single ubiquitin. A distinct subset could not attach the initial ubiquitin, but were able to extend the chain once monoubiquitylation had occurred. It will be interesting to determine whether IAPs also use different E2s to build chains, or whether chain initiation and extension are carried out by one enzyme. As for all RING domains, the mechanism by which they promote ubiquitin transfer has not been revealed, although current models suggest that allosteric activation of the E2 on RING binding is important.57
To prevent uncontrolled protein ubiquitylation, the activity of the RING domain must be tightly regulated, and several studies have suggested that dimerisation of RING domains is a key facet that modulates E3 ligase activity.58 For example, the RING domain of the BRCA1 tumour suppressor protein is activated on heterodimerisation with the RING domain of BARD1.59 Likewise, the RING domains from Ring1b and Bmi form heterodimers, and the E3 ligase activity of Ring1b is increased when Bmi is present.60 Given that the RING domains from IAPs form homo- and heterodimers, it is likely that the E3 ligase activity of IAPs is also modulated by dimerisation. However, IAPs have also been reported to form higher order oligomers,61 and it is uncertain how dimerisation/oligomerisation is regulated in cells.
A regulatory role for domains other than the RING in IAPs is suggested by studies that showed that autoubiquitylation of purified IAPs increased on addition of IAP antagonists.10 This suggests that antagonist binding to BIR2 or BIR3 is conveyed to the RING domain at the C-terminus, promoting autoubiquitylation of IAPs. Clues, as to how this occurs, may lie with the E3 ligase CHIP (C-terminal HSP70 interacting protein), which contains a TPR domain and a U-box domain that is responsible for CHIP's E3 ligase activity. Although the U-box forms a dimer similar to the RING domains, in the crystal structure, only one U-box has an exposed E2-binding site, with the E2-binding site on the other monomer occluded by the TPR domain (Figure 4b).55 Thus, the distant domains influence E2 recruitment and presumably its E3 ligase activity. Furthermore, the C2 domain of the HECT E3 ligase, Smurf2, performs an autoinhibitory function and prevents cycles of futile autoubiquitylation.62 In this case, binding of the adaptor protein, Smad7, to the HECT domain, both relieves inhibition by the C2 domain and facilitates E2 recruitment. Given these examples, it is tempting to invoke a similar mechanism for IAPs, in which antagonist binding interferes with normal interdomain regulation and stimulates autoubiquitylation by the RING domain. Shedding light on the mechanisms of cross-talk between the IAP domains may lead to a much clearer understanding of overall IAP function, and will be key to elucidate how therapeutically promising IAP antagonists achieve their effects at a molecular level.
Conclusions and Future Directions
In recent years, a complex picture of RING-dependent IAP function has emerged. Ubiquitylation of proteins that interact directly or indirectly with IAPs are now the focus of many studies (Figure 3a). Already RIP1 and NIK have been established as key substrates that are brought into the range of IAP E3-ligase activity by adaptor proteins such as TRAFs. Other proteins that directly bind to IAPs, such as MURR1, Mad1 and c-RAF, also undergo IAP-mediated ubiquitylation,63, 64, 65, 66 but both their binding sites on IAPs and the factors that regulate their ubiquitylation remain uncertain. It is likely that additional interacting proteins will be identified, and given the discovery of the UBA domain, it is probable that binding of some of these proteins will be modulated by ubiquitylation. To elucidate the key components of these processes, it will be important to discover whether close association of substrate proteins with IAPs is sufficient to promote their ubiquitylation, or whether there are other mechanisms that regulate this.
The switch between autoubiquitylation and substrate ubiquitylation seems to be key in determining the outcomes of IAP activity (Figure 3b). An important question is how this balance is regulated. Experiments with IBM mimetics and examples in the literature suggest that cross-talk between domains can modulate E3 ligase activity. For example, the BIR domains may interact with the RING domain and restrict E3 ubiquitin ligase activity until the IBM-binding pocket is occupied. The oligomeric state of IAPs may also modulate activity and it will be interesting to see whether this is determined by cellular status.
Clearly, the RING domain is crucial, but to dissect the mechanisms that determine activity analysis of multidomain proteins is likely to be required. As IAP proteins containing more than one domain have proven to be poorly behaved in vitro, success may well depend on combining traditional structural approaches with alternative techniques, such as small-angle X-ray scattering or FRET. Assembling our current knowledge of the separate domains of IAP proteins into a more integrated overview will allow a more complete understanding of how IAPs influence apoptotic pathways and beyond.
Abbreviations
- IAP:
-
inhibitor of apoptosis
- RING:
-
really interesting new gene 1
- BIR:
-
baculovirus IAP repeat
- MDM:
-
mouse double minute
- TNFR:
-
tumour necrosis factor receptor
- IBM:
-
IAP-binding motif
- UBA:
-
ubiquitin associated
- CARD:
-
caspase recruitment domain
- TRAF:
-
TNF receptor-associated factor
- RIP1:
-
receptor-interacting protein 1
- NIK:
-
NF-κB-inducing kinase
- CHIP:
-
C-terminal HSP70 interacting protein
References
Ashkenazi A, Dixit VM . Death receptors: signaling and modulation. Science 1998; 281: 1305–1308.
Youle RJ, Strasser A . The Bcl-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 2008; 9: 47–59.
Salvesen GS, Abrams JM . Caspase activation – stepping on the gas or releasing the brakes? Lessons from humans and flies. Oncogene 2004; 23: 2774–2784.
Crook NE, Clem RJ, Miller LK . An apoptosis-inhibiting baculovirus gene with a zinc finger-like motif. J Virol 1993; 67: 2168–2174.
Uren AG, Coulson EJ, Vaux DL . Conservation of baculovirus inhibitor of apoptosis repeat proteins (BIRPs) in viruses, nematodes, vertebrates and yeasts. Trends Biochem Sci 1998; 23: 159–162.
Riedl SJ, Shi Y . Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol 2004; 5: 897–907.
Vaux DL, Silke J . IAPs, RINGS and ubiquitylation. Nat Rev Mol Cell Biol 2005; 6: 287–297.
Huang H, Joazeiro CA, Bonfoco E, Kamada S, Leverson JD, Hunter T . The inhibitor of apoptosis, cIAP2, functions as a ubiquitin-protein ligase and promotes in vitro monoubiquitination of caspases 3 and 7. J Biol Chem 2000; 275: 26661–26664.
Yang Y, Fang S, Jensen JP, Weissman AM, Ashwell JD . Ubiquitin protein ligase activity of IAPs and their degradation in proteasomes in response to apoptotic stimuli. Science 2000; 288: 874–877.
Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, Kayagaki N, Garg P et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-κB activation, and TNFα-dependent apoptosis. Cell 2007; 131: 669–681.
Vince JE, Wong WW, Khan N, Feltham R, Chau D, Ahmed AU et al. IAP antagonists target cIAP1 to induce TNFα-dependent apoptosis. Cell 2007; 131: 682–693.
Blankenship JW, Varfolomeev E, Goncharov T, Fedorova AV, Kirkpatrick DS, Izrael-Tomasevic A et al. Ubiquitin binding modulates IAP antagonist stimulated proteasomal degradation of c-IAP1 and c-IAP2. Biochem J 2009; 417: 149–160.
Gyrd-Hansen M, Darding M, Miasari M, Santoro M, Zender L, Xue W et al. IAPs contain an evolutionarily conserved ubiquitin-binding domain that regulates NF-κB as well as cell survival and oncogenesis. Nat Cell Biol 2008; 10: 1309–1317.
Hinds MG, Norton RS, Vaux DL, Day CL . Solution structure of a baculoviral inhibitor of apoptosis (IAP) repeat. Nat Struct Biol 1999; 6: 648–651.
Sun C, Cai M, Gunasekera AH, Meadows RP, Wang H, Chen J et al. NMR structure and mutagenesis of the inhibitor-of-apoptosis protein XIAP. Nature 1999; 401: 818–822.
Liu Z, Sun C, Olejniczak ET, Meadows RP, Betz SF, Oost T et al. Structural basis for binding of Smac/DIABLO to the XIAP BIR3 domain. Nature 2000; 408: 1004–1008.
Wu G, Chai J, Suber TL, Wu JW, Du C, Wang X et al. Structural basis of IAP recognition by Smac/DIABLO. Nature 2000; 408: 1008–1012.
Wu JW, Cocina AE, Chai J, Hay BA, Shi Y . Structural analysis of a functional DIAP1 fragment bound to grim and hid peptides. Mol Cell 2001; 8: 95–104.
Yan N, Wu JW, Chai J, Li W, Shi Y . Molecular mechanisms of DrICE inhibition by DIAP1 and removal of inhibition by reaper, hid and grim. Nat Struct Mol Biol 2004; 11: 420–428.
Lu M, Lin SC, Huang Y, Kang YJ, Rich RL, Lo YC et al. XIAP induces NF-κB activation via the BIR1/TAB1 interaction and BIR1 dimerization. Mol Cell 2007; 26: 689–702.
Shiozaki EN, Chai J, Rigotti DJ, Riedl SJ, Li P, Srinivasula SM et al. Mechanism of XIAP-mediated inhibition of caspase-9. Mol Cell 2003; 11: 519–527.
Mastrangelo E, Cossu F, Milani M, Sorrentino G, Lecis D, Delia D et al. Targeting the X-linked inhibitor of apoptosis protein through 4-substituted azabicyclo[5.3.0]alkane Smac mimetics. Structure, activity, and recognition principles. J Mol Biol 2008; 384: 673–689.
Silke J, Kratina T, Chu D, Ekert PG, Day CL, Pakusch M et al. Determination of cell survival by RING-mediated regulation of inhibitor of apoptosis (IAP) protein abundance. Proc Natl Acad Sci USA 2005; 102: 16182–16187.
Uldrijan S, Pannekoek WJ, Vousden KH . An essential function of the extreme C-terminus of MDM2 can be provided by MDMX. EMBO J 2007; 26: 102–112.
Kostic M, Matt T, Martinez-Yamout MA, Dyson HJ, Wright PE . Solution structure of the HDM2 C2H2C4 RING, a domain critical for ubiquitination of p53. J Mol Biol 2006; 363: 433–450.
Linke K, Mace PD, Smith CA, Vaux DL, Silke J, Day CL . Structure of the MDM2/MDMX RING domain heterodimer reveals dimerization is required for their ubiquitylation in trans. Cell Death Differ 2008; 15: 841–848.
Mace PD, Linke K, Feltham R, Schumacher FR, Smith CA, Vaux DL et al. Structures of the cIAP2 RING domain reveal conformational changes associated with ubiquitin-conjugating enzyme (E2) recruitment. J Biol Chem 2008; 283: 31633–31640.
Park HH, Lo YC, Lin SC, Wang L, Yang JK, Wu H . The death domain superfamily in intracellular signaling of apoptosis and inflammation. Annu Rev Immunol 2007; 25: 561–586.
Shi Y . Mechanisms of caspase activation and inhibition during apoptosis. Mol Cell 2002; 9: 459–470.
Chai J, Shiozaki E, Srinivasula SM, Wu Q, Datta P, Alnemri ES et al. Structural basis of caspase-7 inhibition by XIAP. Cell 2001; 104: 769–780.
Huang Y, Park YC, Rich RL, Segal D, Myszka DG, Wu H . Structural basis of caspase inhibition by XIAP: differential roles of the linker versus the BIR domain. Cell 2001; 104: 781–790.
Riedl SJ, Renatus M, Schwarzenbacher R, Zhou Q, Sun C, Fesik SW et al. Structural basis for the inhibition of caspase-3 by XIAP. Cell 2001; 104: 791–800.
Sun C, Cai M, Meadows RP, Xu N, Gunasekera AH, Herrmann J et al. NMR structure and mutagenesis of the third BIR domain of the inhibitor of apoptosis protein XIAP. J Biol Chem 2000; 275: 33777–33781.
Srinivasula SM, Hegde R, Saleh A, Datta P, Shiozaki E, Chai J et al. A conserved XIAP-interaction motif in caspase-9 and Smac/DIABLO regulates caspase activity and apoptosis. Nature 2001; 410: 112–116.
Eckelman BP, Salvesen GS . The human anti-apoptotic proteins cIAP1 and cIAP2 bind but do not inhibit caspases. J Biol Chem 2006; 281: 3254–3260.
Ditzel M, Broemer M, Tenev T, Bolduc C, Lee TV, Rigbolt KT et al. Inactivation of effector caspases through nondegradative polyubiquitylation. Mol Cell 2008; 32: 540–553.
Du C, Fang M, Li Y, Li L, Wang X . Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 2000; 102: 33–42.
Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE et al. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 2000; 102: 43–53.
Yang L, Cao Z, Yan H, Wood W . Coexistence of high levels of apoptotic signaling and inhibitor of apoptosis proteins in human tumor cells: implication for cancer specific therapy. Cancer Res 2003; 63: 6815–6824.
McManus DC, Lefebvre CA, Cherton-Horvat G, St-Jean M, Kandimalla ER, Agrawal S et al. Loss of XIAP protein expression by RNAi and antisense approaches sensitizes cancer cells to functionally diverse chemotherapeutics. Oncogene 2004; 23: 8105–8117.
Arnt CR, Chiorean MV, Heldebrant MP, Gores GJ, Kaufmann SH . Synthetic Smac/DIABLO peptides enhance the effects of chemotherapeutic agents by binding XIAP and cIAP1 in situ. J Biol Chem 2002; 277: 44236–44243.
Guo F, Nimmanapalli R, Paranawithana S, Wittman S, Griffin D, Bali P et al. Ectopic overexpression of second mitochondria-derived activator of caspases (Smac/DIABLO) or cotreatment with N-terminus of Smac/DIABLO peptide potentiates epothilone B derivative-(BMS 247550) and apo-2lTRAIL-induced apoptosis. Blood 2002; 99: 3419–3426.
Vucic D, Fairbrother WJ . The inhibitor of apoptosis proteins as therapeutic targets in cancer. Clin Cancer Res 2007; 13: 5995–6000.
Garber K . New apoptosis drugs face critical test. Nat Biotechnol 2005; 23: 409–411.
Oost TK, Sun C, Armstrong RC, Al-Assaad AS, Betz SF, Deckwerth TL et al. Discovery of potent antagonists of the antiapoptotic protein XIAP for the treatment of cancer. J Med Chem 2004; 47: 4417–4426.
Samuel T, Welsh K, Lober T, Togo SH, Zapata JM, Reed JC . Distinct BIR domains of cIAP1 mediate binding to and ubiquitination of tumor necrosis factor receptor-associated factor 2 and second mitochondrial activator of caspases. J Biol Chem 2006; 281: 1080–1090.
Varfolomeev E, Wayson SM, Dixit VM, Fairbrother WJ, Vucic D . The inhibitor of apoptosis protein fusion c-IAP2.MALT1 stimulates NF-κB activation independently of TRAF1 and TRAF2. J Biol Chem 2006; 281: 29022–29029.
Rothe M, Pan MG, Henzel WJ, Ayres TM, Goeddel DV . The TNFR2-TRAF signaling complex contains two novel proteins related to baculoviral inhibitor of apoptosis proteins. Cell 1995; 83: 1243–1252.
Li X, Yang Y, Ashwell JD . TNF-RII and c-IAP1 mediate ubiquitination and degradation of TRAF2. Nature 2002; 416: 345–347.
Petersen SL, Peyton M, Minna J, Harran P . Autocrine TNFα signaling renders human cancer cells susceptible to Smac-mimetic-induced apoptosis. Cancer Cell 2007; 12: 445–456.
Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell 2008; 30: 689–700.
Vallabhapurapu S, Matsuzawa A, Zhang W, Tseng PH, Keats JJ, Wang H et al. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-κB signaling. Nat Immunol 2008; 9: 1364–1370.
Zarnegar BJ, Wang Y, Mahoney DJ, Dempsey PW, Cheung HH, He J et al. Noncanonical NF-κB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat Immunol 2008; 9: 1371–1378.
Park SM, Yoon JB, Lee TH . Receptor interacting protein is ubiquitinated by cellular inhibitor of apoptosis proteins (c-IAP1 and c-IAP2) in vitro. FEBS Lett 2004; 566: 151–156.
Zhang M, Windheim M, Roe SM, Peggie M, Cohen P, Prodromou C et al. Chaperoned ubiquitylation – crystal structures of the CHIP U box E3 ubiquitin ligase and a CHIP-Ubc13-Uev1a complex. Mol Cell 2005; 20: 525–538.
Christensen DE, Brzovic PS, Klevit RE . E2-BRCA1 RING interactions dictate synthesis of mono- or specific polyubiquitin chain linkages. Nat Struct Mol Biol 2007; 14: 941–948.
Ozkan E, Yu H, Deisenhofer J . Mechanistic insight into the allosteric activation of a ubiquitin-conjugating enzyme by RING-type ubiquitin ligases. Proc Natl Acad Sci USA 2005; 102: 18890–18895.
Knipscheer P, Sixma TK . Protein-protein interactions regulate Ubl conjugation. Curr Opin Struct Biol 2007; 17: 665–673.
Hashizume R, Fukuda M, Maeda I, Nishikawa H, Oyake D, Yabuki Y et al. The RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by a breast cancer-derived mutation. J Biol Chem 2001; 276: 14537–14540.
Buchwald G, van der Stoop P, Weichenrieder O, Perrakis A, van Lohuizen M, Sixma TK . Structure and E3-ligase activity of the RING-RING complex of polycomb proteins Bmi1 and RING1b. EMBO J 2006; 25: 2465–2474.
Rajalingam K, Sharma M, Paland N, Hurwitz R, Thieck O, Oswald M et al. IAP- IAP complexes required for apoptosis resistance of C. trachomatis-infected cells. PLoS Pathog 2006; 2: e114.
Wiesner S, Ogunjimi AA, Wang HR, Rotin D, Sicheri F, Wrana JL et al. Autoinhibition of the HECT-type ubiquitin ligase Smurf2 through its C2 domain. Cell 2007; 130: 651–662.
Burstein E, Ganesh L, Dick RD, van De Sluis B, Wilkinson JC, Klomp LW et al. A novel role for XIAP in copper homeostasis through regulation of MURR1. EMBO J 2004; 23: 244–254.
Dogan T, Harms GS, Hekman M, Karreman C, Oberoi TK, Alnemri ES et al. X-linked and cellular IAPs modulate the stability of c-RAF kinase and cell motility. Nat Cell Biol 2008; 10: 1447–1455.
Tian S, Mewani RR, Kumar D, Li B, Danner MT, Ahmad I et al. Interaction and stabilization of X-linked inhibitor of apoptosis by Raf-1 protein kinase. Int J Oncol 2006; 29: 861–867.
Xu L, Zhu J, Hu X, Zhu H, Kim HT, LaBaer J et al. c-IAP1 cooperates with Myc by acting as a ubiquitin ligase for Mad1. Mol Cell 2007; 28: 914–922.
Acknowledgements
We thank Mark Hinds for critical comments. The work in our laboratory is supported by the Marsden Fund (NZ) and PDM is a recipient of a Health Sciences Career Development Award (University of Otago).
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by G Melino
Rights and permissions
About this article
Cite this article
Mace, P., Shirley, S. & Day, C. Assembling the building blocks: structure and function of inhibitor of apoptosis proteins. Cell Death Differ 17, 46–53 (2010). https://doi.org/10.1038/cdd.2009.45
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2009.45
Keywords
This article is cited by
-
NPD1-mediated stereoselective regulation of BIRC3 expression through cREL is decisive for neural cell survival
Cell Death & Differentiation (2015)
-
The HECTD3 E3 ubiquitin ligase facilitates cancer cell survival by promoting K63-linked polyubiquitination of caspase-8
Cell Death & Disease (2013)
-
X-linked Inhibitor of Apoptosis Protein negatively regulates neuronal differentiation through interaction with cRAF and Trk
Scientific Reports (2013)
-
IAPs regulate the plasticity of cell migration by directly targeting Rac1 for degradation
The EMBO Journal (2012)
-
SMG1 and NIK regulate apoptosis induced by Smac mimetic compounds
Cell Death & Disease (2011)
