
Abstract
Background:
Efficacy and safety are the two considerations when characterising the effects of a new therapy. We sought to apply an innovative method of assessing the benefit–risk balance using data from a completed randomised controlled trial that compared erlotinib vs placebo added to gemcitabine in patients with advanced pancreatic cancer (NCIC CTG PA.3).
Methods:
We applied generalised pairwise comparisons with several prioritised outcome measures (e.g., one or more benefit outcomes and one or more risk outcomes). Here, the first priority outcome was overall survival (OS) time. Differences in OS that exceeded 2 months were considered clinically meaningful. The second priority outcome was toxicity. The overall treatment effect was quantified using the proportion in favour of erlotinib, which can be interpreted as the net proportion of patients who have a better overall outcome with erlotinib as compared with placebo. Sensitivity analyses were performed.
Results:
In this trial 569 patients were randomly assigned in a 1 : 1 ratio to receive gemcitabine plus either erlotinib or a matched placebo. Overall, the method indicated no statistically significant overall treatment effect in favour of erlotinib; if anything, the point estimate of the net proportion leaned in favour of the placebo group (overall proportion in favour of erlotinib=−3.6%, 95% CI, −14.2– 7.1%; P=0.51). The net proportion was never in favour of the erlotinib group throughout all sensitivity analyses.
Conclusions:
Generalised pairwise comparisons make it possible to assess the benefit–risk balance of new treatments using a single statistical test for any number of prioritised outcomes. The benefit–risk assessment was not in favour of adding erlotinib to gemcitabine for the treatment of patients with advanced pancreatic cancer.
Similar content being viewed by others


Main
When characterising a treatment effect, efficacy and safety are the primary considerations. In the reporting of clinical trials, efficacy and safety outcomes are usually reported independently, no formal overall evaluation of the treatment effect is performed (Péron et al, 2012, 2013). Both US Food and Drug Administration and the European Medicines Agency have stressed the importance of a more structured and transparent approach to benefit–risk assessment (BRA) in the evaluation of new therapies (Committee for Medicinal Products for Human Use (CHMP), 2008; Food and Drug Administration, 2011).
Patients with advanced pancreatic cancer have a poor prognosis and the standard first-line regimen is cytotoxic chemotherapy (gemcitabine in monotherapy or in combination with nab-paclitaxel or a combination of 5-fluorouracil, oxaliplatin and irinotecan for patients with good performance status) (Burris et al, 1997; Conroy et al, 2011). The NCIC Clinical Trials Group Study PA.3 (NCIC CTG PA.3) phase III trial investigated the addition of erlotinib to gemcitabine in patients with advanced pancreatic cancer (Moore et al, 2007). Both survival and progression-free survival were significantly better for the combination treatment but the overall benefits were of modest magnitude (HR for overall survival (OS)=0.82, 95% CI, 0.69–0.99; P=0.038). The excess toxicity, the unfavourable cost-effectiveness observed with the combination with erlotinib, (Miksad et al, 2007; Tam et al, 2013) and the absence of a biomarker predictive of erlotinib efficacy, (da Cunha Santos et al, 2010; Boeck et al, 2013) led to a poor uptake of this regimen in the oncology community (Verslype et al, 2007; Saif, 2008; Choi et al, 2012).
No systematic assessment of the benefit–risk balance of erlotinib combination has been performed in the setting of advanced pancreatic cancer. We report here such an assessment based on the method of generalised pairwise comparisons (Buyse, 2010). This method extends the non-parametric Mann–Whitney–Wilcoxon test for a single outcome in the absence of censored data. It allows one to calculate and test the overall benefit of a new treatment based on any number of prioritised outcomes, some reflecting benefit from the intervention (e.g., survival or time to progression) and the others reflecting harm (e.g., treatment-related toxicities and side effects).
Materials and methods
Overview
The NCIC CTG PA.3 trial was an international study that randomised patients with advanced pancreatic cancer to receive gemcitabine in combination with either erlotinib or placebo as first-line treatment. The primary outcome was OS. Progression-free survival (PFS) and toxicity were secondary outcomes.
In this trial, 569 patients were stratified by center, performance status (Eastern Cooperative Oncology Group 0 or 1 vs 2) and extent of disease (locally advanced vs metastatic), and randomly assigned in a 1 : 1 ratio to receive gemcitabine plus either erlotinib or a matched placebo. Progression was evaluated using Response Evaluation Criteria in Solid Tumors (V1.0) every 8 weeks. Toxicity was assessed at every visit using the National Cancer Institute Common Toxicity Criteria version 2.0.
Generalised pairwise comparisons
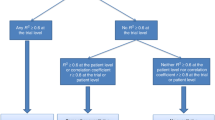
We applied generalised pairwise comparisons extended to several outcome measures (a benefit outcome, and a risk outcome). A full description of generalised pairwise comparisons has been previously published (Buyse, 2010). In brief, pairwise comparisons require consideration of all possible pairs of patients, one taken from the erlotinib arm and the other taken from the placebo arm. Pairwise comparisons are easily stratified for the stratification factors used in the randomisation process. The outcomes of these two patients are compared according to the first priority outcome. The pair is said to be ‘favourable’ if the outcome of the patient in the erlotinib arm is better than the outcome of the patient in the placebo arm, ‘unfavourable’ if the outcome of the patient in the erlotinib arm is worse than the outcome of the patient in the placebo arm and ‘uninformative’ if it cannot be determined which of the two patients has a better outcome (e.g., because of censoring, because the two observations are equal or because the difference of outcomes does not reach a pre-specified threshold value). Such a pairwise comparison is carried out for all pairs of patients, and the difference between the proportion of favourable pairs and the proportion of unfavourable pairs is calculated for the first priority outcome. This difference is called the proportion in favour of treatment for the first priority outcome (Buyse, 2008; Moser and McCann, 2008).
For pairwise comparisons that are uninformative for the first priority outcome, the second priority outcome is used in turn to classify the pair as favourable, unfavourable or uninformative (Table 1). After consideration of the second priority outcome, the ‘overall proportion in favour of treatment’ is calculated to provide an overall assessment of both the benefit and the risks of the treatment, suitably prioritised.
Standard analysis of efficacy and toxicity
A log-rank test adjusted for stratification factors at baseline was used to compare treatment groups in terms of survival. Worst grade adverse events (AE) that were at least possibly related to the study treatment (‘treatment-related AEs’) were reported by treatment group. All analyses were performed on all randomly assigned patients as per the intent-to-treat principle.
Main analysis of the benefit–risk balance
The first priority outcome used in the main analysis was OS. Only pairs of patients with differences in OS exceeding 2 months were considered informative, because smaller differences in OS were not considered clinically meaningful. The second priority outcome was treatment-related AEs, with patients experiencing the lower grade-related AE considered to have had a more favourable outcome. Treatment arms were compared using the overall proportion in favour of the erlotinib group (Δ[erlotinib]). A randomisation test stratified by performance status and extent of disease at diagnosis was performed to test the null hypothesis (H0: Δ[erlotinib]=0). The contribution of each outcome to Δ[erlotinib] was calculated.
Sensitivity analyses
The impact of the choice of outcomes, thresholds and priority on the results was assessed in sensitivity analyses. First, the main analysis was repeated with various thresholds for the minimal OS difference considered as clinically meaningful, ranging from 0 (any difference in OS considered clinically meaningful) to 6 months. Second, the toxicity outcome was defined as a binary variable where only grade ⩾3 AEs were considered. Third, a subgroup analysis was performed among patients treated with 100 mg per day of erlotinib, the actual recommended dose. Finally, a wide range of scenarios integrating OS, PFS and AE grades with several successive thresholds were built to provide a comprehensive assessment of the treatment effects. For each scenario, the overall proportion in favour of the erlotinib group was calculated.
Results
Efficacy outcome
The main analysis of efficacy and safety was conducted after 486 deaths (239 on erlotinib and gemcitabine and 247 on placebo and gemcitabine) and has already been reported (Moore et al, 2007). Overall survival was significantly longer in the erlotinib and gemcitabine arm with an estimated HR of 0.82 (95% CI, 0.69–0.99; P=0.011; log-rank test stratified for performance status, extent of disease). Median survival times were 6.24 months vs 5.91 months for the erlotinib and gemcitabine vs placebo and gemcitabine groups, respectively.
Four hundred and ninety-nine patients had developed progressive disease or had died at the end of the trial. Progression-free survival was significantly longer in the erlotinib and gemcitabine arm with an estimated HR of 0.77 (95% CI, 0.64–0.92; P=0.004; median, 3.75 months vs 3.55 months).
Toxicity outcomes
Two hundred eighty-two patients on the erlotinib and gemcitabine arm and 280 on the placebo and gemcitabine arm received at least one dose of study medication and were available for the assessment of toxicity.
The frequency of all grade and grade ⩾3 treatment-related AEs was higher for the erlotinib and gemcitabine group (90% and 31%, respectively) compared with the placebo and gemcitabine group (76% and 20%, respectively) (Table 2). The increase in grade ⩾3 AEs was especially notable for rash (6% vs 0%).
Benefit–risk assessment
The proportion in favour of the erlotinib group was +4.7%, 95% CI, −5.6–14·6% (thus favouring erlotinib) for the first priority outcome (OS) but −8.3%, 95% CI, −14.2–7.1% (thus favouring placebo) for the second priority outcome (toxicity) among patients uninformative on the OS outcome. Overall, the net proportion favoured non-significantly the placebo group (overall Δ[erlotinib]=−3.6, 95% CI, −14.2– 7.1; P=0.51), suggesting an unfavourable benefit–risk balance of erlotinib added to gemcitabine (Table 3).
Sensitivity analyses
The analysis was repeated with various values for the OS threshold, varying between 0 and 6 months. When the OS threshold was set at 0 month, meaning that any difference in OS was considered meaningful, the overall analysis was not statistically significant (overall proportion in favour of erlotinib=2.3, 95% CI, −8.1–12.7; P=0.67). This setting gave a large weight to the first priority OS outcome, because any survival improvement was considered clinically significant, regardless of AEs. As the OS threshold increased, the overall assessment leaned more and more in favour of the placebo group. It reached statistical significance in favour of erlotinib for values of the OS threshold >5 months (Figure 1).

Benefit–risk of erlotinib according to the minimum survival benefit considered clinically meaningful. Proportion in favour of erlotinib according to the minimum survival benefit considered clinically meaningful. First priority outcome: overall survival. Second priority outcome: worst grade of at least possibly related adverse events. Solid black line with asterisks: proportion in favour of erlotinib according to the first priority outcome (OS) only. Solid light-grey line with points: overall proportion in favour of erlotinib.
The analysis was repeated using a threshold of two AE grades for the second priority toxicity outcome (hence, in this analysis, a difference of one grade or less was not considered clinically meaningful). Again, the analysis tended to favour the placebo group but remained non-significant statistically (Table 4).
When only Grade ⩾3 AEs were considered in the second priority toxicity outcome, the overall proportion in favour of erlotinib was again low for OS threshold under 2 months (+1.5, 95% CI, −8.5–11.4; P=0.77) and became negative for OS thresholds larger than 2.5 months (Figure 2). The analyses never reached statistical significance for the tested OS thresholds (up to 6 months).

Benefit–risk of erlotinib according to the minimum survival benefit considered clinically meaningful. Only grade 3–4 adverse events are included in this analysis. Proportion in favour of erlotinib according to the minimum survival benefit considered clinically meaningful. First priority outcome: overall survival. Second priority outcome: presence of a grade 3–4 at least possibly related adverse events. Solid black line with asterisks: proportion in favour of erlotinib according to the first priority (OS) outcome only. Solid light-grey line with points: overall proportion in favour of erlotinib.
When skin rashes were excluded from the list of AEs analyzed in the second priority outcome, the overall analysis was not in favour of erlotinib (overall proportion in favour of erlotinib=−0.3, 95% CI, −9.1–8.4; P=0.94) (Table 5). A subgroup analysis was performed according to the occurrence of a grade ⩾2 rash in the erlotinib group. The benefit–risk of erlotinib in the subgroup of patients experiencing grade ⩾2 rashes was statistically significantly favourable (Δ[erlotinib]=13.7; P=0.032), and it was statistically significantly unfavourable in the subgroup of patients with grade 0 or 1 rashes (Δ[erlotinib]=−13.8; P=0.016) (Table 6).
In the subgroup of the 521 patients treated with 100 mg per day of erlotinib, the main analysis of benefit–risk once again was not in favour of the erlotinib (overall proportion in favour of erlotinib=−2.7, 95% CI, −13.6–8.1; P=0.62).
Comprehensive sensitivity analyses of the benefit–risk were carried out using various thresholds for OS, PFS and worst AE grade. Some scenarios with clinically meaningful choices of end point prioritisation and of thresholds are presented in Table 7. For none of the scenarios considered was the overall benefit risk assessment in favour of erlotinib.
Discussion
We have used generalised pairwise comparisons, prioritised on several outcomes, to perform an assessment of the benefit–risk balance of adding erlotinib to gemcitabine for the treatment of patients with advanced pancreatic cancer. These analyses showed that the OS benefit in favour of erlotinib diminished when using increased thresholds for the OS benefit and/or adding AEs in an assessment of the net benefit of the combination. The benefit risk assessment did not favour adding erlotinib in the main analysis, and this result was confirmed in all sensitivity analyses.
The method of generalised pairwise comparisons gives higher priority to the outcome considered clinically more important – in this case, overall survival was considered more important than any grade of toxicity. The method can incorporate both a priority and a threshold for each of the outcomes considered (in this instance, OS and treatment-related toxicities), and as such it reflects the thinking process of clinicians and decision makers, who try to assess the net effect of a new treatment on several outcomes considered to be of clinical importance. As such, the method may be particularly informative in health technology assessment.
Several methods have been proposed to help the scientific assessment of the benefit–risk balance of interventions. These methods are most frequently designed to weigh relevant efficacy and safety data into a single construct (Committee for Medicinal Products for Human Use (CHMP), 2008). QALY is a measurement of health status that assigns a weight in each period of time according to the quality of life during this period (Weinstein et al, 2009). It might be used to adjust a gain in survival to an increased level of toxicity by assigning a smallest weight to the time of survival with significant toxicity. However, it requires clearly defined health states, as well as weights for each state, which might be difficult to establish when planning a trial. This limitation makes QALY difficult to use as a primary end point to evaluate therapeutic interventions, and a more suitable tool for medico-economic evaluation (Whitehead and Ali, 2010). Other methods such as Overall Treatment Utility (OTU) can be used to combine subjective and objective measures of the treatment effect into a single composite end point. However the respective weights of the different treatment effects included in OTU may be difficult to justify and to report (Seymour et al, 2011).
The method of generalised pairwise comparisons only requires the priority of each outcome to be defined. Sensitivity analyses are useful to confirm the conclusion of the main analysis. Indeed, the conclusion may rest entirely on arbitrary (though arguably relevant) choices made regarding outcome priorities and thresholds values (if any). Most clinicians and patients would agree that small gains in survival cannot be considered as a positive outcome if such gains are obtained at the expense of severe toxicities. However, determining the minimal survival benefit threshold for which most patients would accept to experience a treatment-related AE is very complex. It may depend on the type of AE and its grade, and it may vary considerably from patient to patient. Survival benefits may be offset by severe and/or long-term AEs. Investigators can now use generalised pairwise comparisons to test the benefit–risk balance of investigational therapies, depending on the level of tolerable toxicity that is deemed acceptable for a given magnitude of survival benefit. Various scenarios for the threshold of survival benefit and the grades of AEs are reported in the Table 7. Throughout all the scenarios, the benefit–risk balance leaned against erlotinib, which does provide some confirmation of the results of the main analysis. Moreover the clinical impact of AEs may vary a lot depending of the type of AEs, even among AEs of the same grade. When skin rashes were excluded from the list of relevant adverse events, the benefit risk assessment of erlotinib was close to zero.
Relevant toxicity criteria could potentially vary from trial to trial. For example, a risk assessment could focus on predefined AEs of special interest, on all severe AEs, on severe treatment-related AEs, or on AEs leading to drug discontinuation. (Ioannidis et al, 2004) For the PA.3 trial, the frequency of lethal AEs or of AEs leading to treatment discontinuation was low, as well as the frequency of grade 3–4 AEs.
Generalised pairwise comparisons are useful to perform a quantitative assessment of the benefit–risk balance of a new treatment as compared with a standard therapy. Such an assessment is especially useful when overall efficacy differences are small, and no subset of patients has been identified as being more likely to benefit from treatment. In such cases, generalised pairwise comparisons provide a clinically intuitive way of comparing patients with respect to all important efficacy and toxicity outcomes, with full flexibility as to the priority of each outcome, and a threshold of clinical significance. In particular, when some patients benefit from treatment at the price of a given toxicity (e.g., severe treatment-related rash after administration of a tyrosine kinase inhibitor), the prioritisation of their outcomes naturally ensures that the benefit trumps the toxicity in the overall assessment of the benefit–risk balance.
Change history
17 March 2015
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Boeck S, Jung A, Laubender RP, Neumann J, Egg R, Goritschan C, Ormanns S, Haas M, Modest DP, Kirchner T, Heinemann V (2013) KRAS mutation status is not predictive for objective response to anti-EGFR treatment with erlotinib in patients with advanced pancreatic cancer. J Gastroenterol 48: 544–548.
Burris HA, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo AM, Tarassoff P, Nelson R, Dorr FA, Stephens CD, Von Hoff DD (1997) Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol 15: 2403–2413.
Buyse M (2008) Reformulating the hazard ratio to enhance communication with clinical investigators. Clin Trials 5: 641–642.
Buyse M (2010) Generalized pairwise comparisons of prioritized outcomes in the two-sample problem. Stat Med 29: 3245–3257.
Choi M, Razzaque S, Kim R (2012) Systemic therapy of advanced pancreatic cancer: has the landscape changed? Clin Adv Hematol Oncol 10: 442–451.
Committee for Medicinal Products for Human Use (CHMP) (2008) Report of the CHMP working group on benefit-risk assessment models and methods http://www.ema.europa.eu Last accessed March 2014.
Conroy T, Desseigne F, Ychou M (2011) FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 364: 1817–1825.
Da Cunha Santos G, Dhani N, Tu D, Chin K, Ludkovski O, Kamel-Reid S, Squire J, Parulekar W, Moore MJ, Tsao MS (2010) Molecular predictors of outcome in a phase 3 study of gemcitabine and erlotinib therapy in patients with advanced pancreatic cancer: National Cancer Institute of Canada Clinical Trials Group Study PA.3. Cancer 116: 5599–5607.
Food and Drug Administration (2011) PDUFA Reauthorization performance goals and procedures fiscal years 2013 through 2017. [Internet] http://www.fda.gov/downloads/ForIndustry/User-Fees/PrescriptionDrugUserFee/UCM270412.pdf Last accessed March 2014.
Ioannidis JPA, Evans SJW, Gøtzsche PC, O’Neill RT, Altman DG, Schulz K, Moher D CONSORT Group (2004) Better reporting of harms in randomized trials: an extension of the CONSORT statement. Ann Intern Med 141: 781–788.
Miksad RA, Schnipper L, Goldstein M (2007) Does a statistically significant survival benefit of erlotinib plus gemcitabine for advanced pancreatic cancer translate into clinical significance and value? J Clin Oncol 25: 4506–4507, author reply 4508.
Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, Campos D, Lim R, Ding K, Clark G, Voskoglou-Nomikos T, Ptasynski M, Parulekar W National Cancer Institute of Canada Clinical Trials Group (2007) Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 25: 1960–1966.
Moser BK, McCann MH (2008) Reformulating the hazard ratio to enhance communication with clinical investigators. Clin Trials 5: 248–252.
Péron J, Maillet D, Gan HK, Chen EX, You B (2013) Adherence to CONSORT adverse event reporting guidelines in randomized clinical trials evaluating systemic cancer therapy: a systematic review. J Clin Oncol 31: 3957–3563.
Péron J, Pond GR, Gan HK, Chen EX, Almufti R, Maillet D, You B (2012) Quality of reporting of modern randomized controlled trials in medical oncology: a systematic review. J Natl Cancer Inst 104: 982–989.
Saif MW (2008) Is there a standard of care for the management of advanced pancreatic cancer? Highlights from the Gastrointestinal Cancers Symposium, Orlando, FL, USA. January 25-27, 2008. JOP 9: 91–98.
Seymour MT, Thompson LC, Wasan HS, Middleton G, Brewster AE, Shepherd SF, O'Mahony MS, Maughan TS, Parmar M, Langley RE FOCUS2 Investigators National Cancer Research Institute Colorectal Cancer Clinical Studies Group (2011) Chemotherapy options in elderly and frail patients with metastatic colorectal cancer (MRC FOCUS2): an open-label, randomised factorial trial. Lancet 377: 1749–1759.
Tam VC, Ko YJ, Mittmann N, Cheung MC, Kumar K, Hassan S, Chan KK (2013) Cost-effectiveness of systemic therapies for metastatic pancreatic cancer. Curr Oncol 20: e90–e106.
Verslype C, Van Cutsem E, Dicato M, Cascinu S, Cunningham D, Diaz-Rubio E, Glimelius B, Haller D, Haustermans K, Heinemann V, Hoff P, Johnston PG, Kerr D, Labianca R, Louvet C, Minsky B, Moore M, Nordlinger B, Pedrazzoli S, Roth A, Rothenberg M, Rougier P, Schmoll HJ, Tabernero J, Tempero M, van de Velde C, Van Laethem JL, Zalcberg J (2007) The management of pancreatic cancer. Current expert opinion and recommendations derived from the 8th World Congress on Gastrointestinal Cancer, Barcelona, 2006. Ann Oncol 18: 1–10.
Weinstein MC, Torrance G, McGuire A (2009) QALYs: the basics. Value Health 12: 5–9.
Whitehead SJ, Ali S (2010) Health outcomes in economic evaluation: the QALY and utilities. Br Med Bull 96: 5–21.
Acknowledgements
Dr Julien Péron is the recipient of a grant from the Nuovo-Soldati Research Foundation. The study was not funded.
Author contributions
Julien Péron and Marc Buyse: design of the study, data collection and analysis, and writing and approval of the manuscript. Guarantor: Pascal Roy and Laurent Roche: design of the study, data analysis, and writing and approval of the manuscript. Keyue Ding and Wendy R Parulekar: data collection and analysis and writing and approval of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Péron, J., Roy, P., Ding, K. et al. Assessing the benefit–risk of new treatments using generalised pairwise comparisons: the case of erlotinib in pancreatic cancer. Br J Cancer 112, 971–976 (2015). https://doi.org/10.1038/bjc.2015.55
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2015.55
Keywords
This article is cited by
-
Composite endpoints, including patient reported outcomes, in rare diseases
Orphanet Journal of Rare Diseases (2023)
-
Standards de chimiothérapie, perspectives et thérapies ciblées dans l’adénocarcinome du pancréas
Oncologie (2015)
