
Abstract
Aim:
To investigate the inhibitory interactions of bufalin and CYP3A4.
Methods:
Recombinant human CYP3A4 was incubated with bufalin in vitro. Bufalin was administered ig and iv to Wistar rats to further estimate its impact on CYP3A4, and midazolam was given to index the activity of CYP3A4.
Results:
The IC50 of bufalin was 14.52 μmol/L. Bufalin affected CYP3A4 activity with increases in AUC0–t and t1/2, and decreases in CL and the formation of 1-hydroxy-midazolam after ig or iv administration of midazolam (P<0.05). An increase in Cmax after ig bufalin administration (P<0.05) was observed.
Conclusion:
Bufalin showed a modest but significant inhibition of CYP3A4 both in vitro and in vivo. The likelihood of an interaction between bufalin and the CYP3A4-metabolized drugs in human might not be negated.
Similar content being viewed by others
Introduction
Human cytochrome P450 3A4 (CYP3A4), a member of the cytochrome P450 mixed-function oxidase system, is one of the most important enzymes involved in the metabolism of xenobiotics in the body. CYP3A4 accounts for roughly 40% of the total cytochrome P450 in human liver microsomes, and metabolizes more than 50% of the clinically used drugs. CYP3A4 is also expressed in the small intestine, where it contributes to the first-pass elimination of many drugs1, 2, 3, 4.
The ability of CYP3A4 to metabolize numerous structurally unrelated compounds accounts for the large number of documented drug interactions associated with CYP3A4 inhibition. These interactions and subsequent CYP3A4 inhibition can lead to serious adverse actions, especially when other drugs are metabolized by the same enzyme, which inevitably results in the reduced clearance or increased absorption of the affected drugs. According to the FDA DDI Guidance, in vitro experiments are routinely performed to determine whether a promising new chemical entity inhibits CYP3A4, and thus alters the safety and efficacy profile of the concomitant drugs or their active metabolites in important ways if they are also substrates of the enzyme.
Interactions exist not only between different drugs, but also between drugs and dietary supplements. Alternative medicines, including traditional Chinese medicines, are classified as dietary supplements.
Under the Dietary Supplement and Education Act of 1994, dietary supplement manufacturers are not required to evaluate the potential for drug interactions, but adverse effects, toxicity and even death have been reported from using dietary supplements5, 6. According to surveys conducted in the United States, a considerable proportion of the population is believed to use botanical dietary supplements together with prescribed drugs. In Asian countries, a strong practice of traditional medicine exists, especially when Western medicines do not provide the desired results, and adverse drug interactions can go unrecognized or misconstrued as part of the healing process. This is a particular concern among the elderly generation, who typically take multiple prescription medications7, 8, 9. All of these facts indicate the prevalent nature of drug interactions associated with CYP3A4 and justify the present investigation of the effect of bufalin on this particular enzyme.
Bufalin is the major digoxin-like immunoreactive component of Chan Su, a traditional Chinese medicine obtained from the skin and parotid venom glands of the toad9, 10, 11, 12, 13, 14, 15. Chan Su is the major component of such popular traditional Chinese herbal remedies as Liu-shen-wan, She-xiang-bao-xin-wan and Kyushin13, 16. These traditional Chinese herbal remedies have been widely applied in China and other Asian countries, and are currently used as alternatives to Western treatments. Bufalin is known to increase vasoconstriction, vascular resistance and blood pressure, and has been shown to induce apoptosis in human leukemia HL-60 cells, endometriotic stromal cells, and prostate cancer cells. In terms of its anti-tumor activity, bufalin has been demonstrated to inhibit the growth of tumor cells by inducing apoptosis and cell cycle arrest12, 13, 14, 15, 16, 17, 18.
Few studies have examined the interaction of bufalin with prescription medications, and none have been conducted on its impact on the CYP3A4 enzyme system. Since bufalin has shown inhibitory effects on recombinant CYP3A4 (rCYP3A4) among the five investigated bufadienolides (cinobufagin, resibufogenin, bufalin, bufotalin and arenobufagin) in our preliminary tests, the effect of bufalin on CYP3A4 was further evaluated both in vitro and in vivo in the present experiments.
Materials and methods
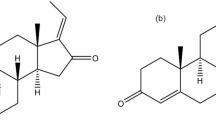
rCYP3A4, glucose 6-phosphate, 1-hydroxy-midazolam, diazepam, NADPH and glucose 6-phosphate dehydrogenase were from Sigma (St Louis, MO, USA), midazolam was from Jiangsu Nhua Pharmaceutical Co (Nanjing, China), tinidazole was from the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China), and K2HPO4, NaOH, and MgCl2·6H2O were from Sinopharm Chemical Reagent Co, Ltd (Shanghai, China). Bufalin (for chemical structure, see Figure 1) was prepared in-house with a purity of 99.0% by HPLC analysis. Its chemical structure was identified based on chemical reactions and spectral analysis (1H NMR, 13C NMR, 2D NMR, MS, UV, and IR).

Chemical structure of bufalin.
HPLC-grade acetonitrile was from Caledon Laboratories Ltd (Georgetown, Ont, Canada). HPLC-grade formic acid was from Tedia Company Inc (Fairfield, OH, USA). Ultra pure water was obtained from a Milli-Q PLUS purification system (Millipore, MA, USA). All other reagents were of analytical grade.
Sample preparation Precipitated samples from in vitro experiments were centrifuged at 10 000×g for 5 min, and the supernatants were analyzed by LC/MS/MS for the amount of metabolite (1-hydroxy-midazolam) formed. The percent of metabolic activity was calculated by comparing with the enzyme activity in the control samples that did not contain bufalin. Each set of incubations was carried out with four control samples.
To 100 μL of a plasma sample, 20 μL of acetonitrile and 240 μL of diazepam (IS for in vivo assessment) were added. After centrifugation at 4000×g for 5 min, 10 μL of the resulting supernatant was injected into the LC/MS/MS system to determine the concentrations of midazolam.
Standard stock solutions of tinidazole (IS for in vitro assessment), diazepam and 1-hydroxy-midazolam were prepared in acetonitrile with final concentrations of 1, 10, and 100 μg/mL, respectively. Tinidazole and diazepam were then diluted into working solutions with concentrations of 1 μg/mL and 5 ng/mL, respectively. The calibration curves ranged from 0.5 to 2000 ng/mL (samples collected at times 0.083, 0.167, 0.33, 0.5 h after iv administration were diluted to within the linear range) for midazolam, and 1 to 1000 ng/mL for 1-hydroxy-midazolam. The limits of quantification were 1 and 0.2 ng/mL for 1-hydroxy-midazolam and midazolam, respectively. All analytical results were within acceptance criteria.
Study design
In vitro assessment of IC50 with rCYP3A4 activity rCYP3A4 (50 pmol) was incubated with bufalin in 100 mmol/L potassium phosphate buffer (pH 7.4) with 5 μmol/L MgCl2 and an NADPH-generating system consisting of 10 μmol/L glucose 6-phosphate, 1 μmol/L NADPH, and 2.5 U/mL glucose 6-phosphate dehydrogenase in a total volume of 0.2 mL. Incubations were carried out in a 37 °C shaking water bath for 5 min. Reactions were stopped by adding 0.4 mL of ice-cold acetonitrile containing 1 μg/mL tinidazole.
The IC50 value of bufalin was calculated from dose-response curves consisting of 11 points (0, 0.1, 0.5, 1, 2.5, 5, 7.5, 10, 15, 20, and 30 μmol/L) per duplicate that were reproduced in three independent experiments.
Assessment of in vivo CYP3A4 activity CYP3A4 activity was assessed in vivo with the principle that midazolam was a substrate for CYP3A4, and that the activity of this enzyme was inversely reflected in the plasma levels of ig- or iv-administered midazolam.
Clean grade male Wistar rats (Shanghai SLAC Lab Animal Co Ltd, Shanghai, China) that were 6 weeks old and weighed 210±20 g were allowed to acclimate to the animal house facility of the Second Military Medical University for at least 3 days prior to the experiments. The rats were maintained on a 12 h light/dark cycle with laboratory rodent chow and tap water ad libitum, except during the fasting periods prior to dosing, when all food was removed. These animals were fasted until 4 h post-dose.
Rats were randomly divided in four groups (n=6). Groups 1 and 2 were administered ig at 10 mg/kg of bufalin, while groups 3 and 4 received vehicle control (0.5% sodium carboxymethyl cellulose) in comparable amounts. Midazolam was dosed ig at 10 mg/kg 1 h after the administration of bufalin or the vehicle control to groups 1 and 3, and groups 2 and 4 were dosed iv at 5 mg/kg 0.25 h after the administration of bufalin or the vehicle control. After a 12 h fast, the animals were dosed as described above. Heparinized blood samples were collected at 5, 10, 15, 30 min and 1, 1.5, 2, 3, 4, 6, and 9 h post-dose, then immediately centrifuged at 10 000×g at room temperature for 5 min. Plasma was removed and stored at -20 °C until analysis.
All animal experiments were approved by the Institutional Animal Care and Use Committee of the Second Military Medical University.
Data analysis
Pharmacokinetic analysis All pharmacokinetic parameters were determined by non-compartmental analysis. The peak plasma level (Cmax) and the time to reach the peak plasma concentration (tmax) were obtained directly from the concentration-time data. The elimination rate constant (Ke) was calculated from the slope of the logarithm of the plasma concentration versus time using the final four points. The apparent elimination half-time (t1/2) was calculated as 0.693/Ke. The area under the plasma concentration-time curve (AUC) and the area under the first moment curve (AUMC) were calculated using the trapezoidal rule. Total body clearance (CL) was calculated as X0/AUC. The mean residence time (MRT) was calculated by dividing AUMC by AUC. Each value was expressed as the mean±SD.
Statistic analysis The Student's t-test at 95% confidence (P<0.05) was performed on data from the bufalin and control groups to determine any significant differences between the means of the different treatment groups.
Apparatus and chromatographic conditions An Agilent 6410 triple quadrupole LC/MS/MS system (Agilent Corporation, MA, USA) with the MassHunter workstation software (Agilent Corporation, MA, USA) was used to perform the analytical procedures. The system consisted of a G1311A quaternary pump, G1322A vacuum degasser, G1329A autosampler, and G1316A thermal column compartments.
The separation of the plasma samples was performed on a ZORBAX SB-C18 column (3.5 μm, 2.1×100 mm, Agilent Corporation, MA, USA) and a C18 guard column (5 μm, 4.0×2.0 mm, Phenomenex, CA, USA) with an isocratic elution of acetonitrile/0.1% formic acid (40:60, v/v) at a flow rate of 0.3 mL/min. The column was maintained at 35 °C and the injection volume was 10 μL.
Ionization was achieved using the electrospray method in positive mode with the spray voltage set at 4000 V. Nitrogen was used as nebulizer gas and the nebulizer pressure was set at 40 psi with a source temperature of 105 °C. Desolvation gas (nitrogen) was heated to 350 °C and delivered at a flow rate of 10 L/min. For collision-induced dissociation (CID), high-purity nitrogen was used as collision gas at a pressure of 0.1 MPa. Quantification was preformed using multiple reaction monitoring (MRM) of m/z 326.1→291.3 for midazolam, m/z 285.1→193.1 for diazepam, m/z 342.2→324.2 for 1-hydroxy-midazolam and m/z 248.2→121.1 for tinidazole. The fragment energies of MS1 for midazolam, diazepam, 1-hydroxy-midazolam and tinidazole were set at 220, 180, 160, and 140 V, respectively. Optimized collision energies of 30, 31, 20, and 15 were used for midazolam, diazepam, 1-hydroxy-midazolam and tinidazole, respectively. The peak widths of the precursor and product ions were maintained at 0.7 amu at half-height in the MRM mode.
Results
Mass spectrometric detection
The full-scan spectra showed that predominant protonated molecular ions at m/z 326.1, 285.1, 342.2, and 248.2, which were chosen as parent ions for fragmentation in the multiple-reaction-monitoring (MRM) mode, were produced from midazolam, diazepam, 1-hydroxy-midazolam and tinidazole, respectively. The daughter spectra of the parent ions revealed that the predominant daughter fragments were m/z 291.3, 193.1, 324.2, and 121.1, respectively, and these product ions were chosen for analyte quantitation.
IC50 for bufalin with rCYP3A4
The activity of rCYP3A4 was measured in the presence of various bufalin concentrations. The IC50 value for bufalin was determined to be 14.52 μmol/L, and the representative inhibition profile is shown in Figure 2.

Representative inhibition profiles for rCYP3A4 activities by bufalin.
Pharmacokinetics of midazolam in bufalin-treated rats
All pharmacokinetic parameters of ig and iv administered midazolam and the 1-hydroxy-midazolam formed were calculated using the non-compartmental pharmacokinetic model. The resulting concentration–time profile is shown in Figure 3, and the pharmacokinetic parameter values for midazolam and 1-hydroxy-midazolam are summarized in Table 1 and Table 2, respectively.

(A) Mean plasma concentration-time curve of midazolam after its iv administration (5 mg/kg) to bufalin-(10 mg/kg, ig)/vehicle-treated rats; (B) Mean plasma concentration-time curve of midazolam after its ig administration (10 mg/kg) to bufalin-(10 mg/kg, ig)/vehicle-treated rats; (C) Mean plasma concentration-time curve of 1-hydroxy midazolam after iv administration of midazolam (5 mg/kg) to bufalin-(10 mg/kg, ig)/vehicle-treated rats; (D) Mean plasma concentration-time curve of 1-hydroxy midazolam after ig administration of midazolam (10 mg/kg) to bufalin-(10 mg/kg, ig)/vehicle-treated rats (n=6).
The Student's t-test revealed a significant influence of treatment of bufalin on the ig pharmacokinetics of midazolam. When compared with the control group, the AUC0–t, AUC0–∞, t1/2, and Cmax in the bufalin-treated group were increased (P=0.01, 0.01, 0.04, 0.02). Similarly, bufalin treatment also had a significant influence on the pharmacokinetics of iv administered midazolam. The iv AUC0–t, AUC0–∞, and t1/2 in the bufalin-treated group were increased (P=0.02, 0.02, 0.04), while the CL was reduced (P=0.009). Changes in the pharmacokinetic values of 1-hydroxy-midazolam were also investigated, and a reduction in AUC0–t and AUC0–∞ after iv and ig administration were observed (P=0.02, 0.02 for iv, and P=0.04, 0.04 for ig).
Discussion
There has been a steady increase in the use of dietary supplements during the last decade in the United States, and the availability of Chinese medicines is no longer limited to the Asian population. Chan Su is one of these medicines. One of the major active ingredients of Chan Su, bufalin, is known to exhibit a variety of biological activities. Bufalin has been shown to induce apoptosis in human leukemia HL-60 cells and human tumor cells12, 13, 14, 15, 16, 17, 18. In preliminary experiments, bufalin has shown modest inhibition on hepatic CYP3A4. Thus, in vitro and in vivo experiments were carried out to further estimate its inhibitory effect on the enzyme.
After in vitro incubation of bufalin with rCYP3A4, a modest inhibitory effect on the formation of 1-hydroxy-midazolam was seen, and the IC50 for bufalin was determined. However, the interpretation of the in vitro assay data and, in particular, its application to risk assessment, is not straightforward. To further investigate its impact on the CYP3A4 enzyme system, an in vivo protocol for CYP3A4 inhibition by bufalin was designed. As trade secrets, the exact content of Chan Su in herbal remedies such as Liu-shen-wan and She-xiang-bao-xin-wan is not known, so a single dose of 10 mg/kg was selected to avoid any adverse effects in the short term and to cover the total amount of multiple administrations within a day. The in vivo experiment showed statistically significant differences between values of the parameters obtained from rats administered with bufalin and those administered vehicle, which indicates a potential interaction when bufalin is consumed with drugs that are metabolized by CYP3A4. Additional risk would arise when the drug involved has a narrow therapeutic index.
CYP3A4 and CYP3A5 are the major isoforms of CYP3A expressed in adults. The expression of CYP3A5 was found to be very low or at undetectable levels in 70% to 75% of livers and small intestines examined19, 20. However, CYP3A4 was found to be expressed in all adult livers21 and to be the predominant CYP3A expressed in human enterocytes, indicating that the contribution of CYP3A5 to midazolam 1-hydroxylation is small22, 23. In spite of the high similarity (84% of identity in amino acid sequence) and substantial overlap of substrate specificities3, 24, the CYP3A4 and CYP3A5 proteins have been shown to possess different enzymatic properties, including susceptibility to inhibitors. A previous study showed that the catalytic activity of CYP3A5 was less than that of CYP3A4. The inhibitory potency of ketoconazole toward rCYP3A5 was about 5 to 19 times lower than rCYP3A4 and human liver microsomes for midazolam, triazolam, nifedipine, and testosterone24. All of the above suggested a less important role for CYP3A5 compared with CYP3A4 in potential drug-drug interactions, even if bufalin did inhibit CYP3A5. Although effects on the function of P-glycoprotein (MDR) may also be important when considering the mechanism of the changes in the pharmacokinetics of orally administered drugs, it can be excluded in this case, since midazolam has been reported not to be a substrate for P-glycoprotein6, 25. The facts described above indicated that CYP3A4 inhibition in the presence of bufalin was a major cause of the changes in the pharmacokinetics of midazolam.
In conclusion, the present study demonstrates that bufalin, which has been reported to exhibit significant anti-tumor activity12, 13, 14, 15, 16, 17, 18, showed a modest but significant inhibition of CYP3A4 both in vitro and in vivo, which was confirmed by bioequivalence evaluation. The likelihood of an in vivo interaction between bufalin and CYP3A4-metabolized drugs in humans might not be negated, although more data on multiple-dose pharmacokinetics should be collected to complete the inhibitory profile, and further experiments need to be carried out to determine whether bufalin could be metabolized by the CYP3A4 system or whether its metabolites could inhibit the enzyme. The clinical significance of these findings could be assessed by follow-up studies in other animals, and eventually in humans.
Author contribution
Wen XU designed the research; Hai-yun LI performed the research; Wei-dong ZHANG contributed new reagents or analytical tools; Hai-yun LI analyzed data; Hai-yun LI wrote the paper, Wen XU, Xi ZHANG, Wei-dong ZHANG and Li-wei HU revised the paper.
References
Luo G, Cunningham M, Kim S, Burn T, Lin J, Sinz M, et al. CYP3A4 induction by drugs: correlation between a pregnane X receptor reporter gene assay and CYP3A4 expression in human hepatocytes. Drug Metab Dispos 2002; 30: 795–804.
Wang RW, Newton DJ, Liu N, Atkins WM, Lu AY . Human cytochrome P-450 3A4: in vitro drug drug interaction patterns are substrate-dependent. Drug Metab Dispos 2000; 28: 360–6.
Quintieri L, Palatini P, Nassi A, Ruzza P, Floreani M . Flavonoids diosmetin and luteolin inhibit midazolam metabolism by human liver microsomes and recombinant CYP3A4 and CYP3A5 enzymes. Biochem Pharmacol 2008; 75: 1426–37.
Casabar RC, Wallace AD, Hodgson E, Rose RL . Metabolism of endosulfan-α by human liver microsomes and its utility as a simultaneous in vitro probe for CYP2B6 and CYP3A4. Drug Metab Dispos 2006; 34: 1779–85.
Yetley E . Multivitamin and multimineral dietary supplements: definitions, characterization, bioavailability, and drug interactions. Am J Clin Nutr 2007; 85: 269S–76S.
Bick RJ, Poindexter BJ, Sweney RR, Dasgupta A . Effects of Chan Su, a traditional Chinese medicine, on the calcium transients of isolated cardiomyocytes: cardiotoxicity due to more than Na, K-ATPase blocking. Life Sci 2002; 72: 699–709.
Nishikawa M, Ariyoshi N, Kotani A, Ishii I, Nakamura H, Nakasa H, et al. Effects of continuous ingestion of green tea or grape seed extracts on the pharmacokinetics of midazolam. Drug Metab Pharmacokinet 2004; 19: 280–9.
Shimazaki M, Martin J . Do herbal agents have a place in the treatment of sleep problems in long-term care? J Am Med Dir Assoc 2007; 8: 248–52.
Delgoda R, Westlake A . Herbal interactions involving cytochrome P450 enzymes. Toxicol Rev 2004; 23: 239–49.
Bhuiyana B, Fantb M, Dasgupta A . Study on mechanism of action of Chinese medicine Chan Su: dose dependent biphasic production of nitric oxide in trophoblastic BeWo cells. Clin Chim Acta 2003; 330: 179–84
Biddle DA, Datta P, Wells A, Dasgupta A . Falsely elevated serum digitoxin concentrations due to cross-reactivity of water-extractable digitoxin-like immunoreactivity of Chinese medicine Chan SU: elimination of interference by use of a chemiluminescent assay. Clin Chim Acta 2000; 300: 151–8.
Yin JQ, Shen JN, Su WW, Wang J, Huang G, Jin S, et al. Bufalin induces apoptosis in human osteosarcoma U-2OS and U-2OS methotrexate300-resistant cell lines. Acta Pharmacol Sin 2007; 28: 712–20.
Nasu K, Nishida M, Ueda T, Takai N, Bing S, Narahara H, et al. Bufalin induces apoptosis and the G0/Gl cell cycle arrest of endometriotic stromal cells: a promising agent for the treatment of endometriosis. Mol Hum Reprod 2005; 11: 817–23.
Zhang L, Nakaya K, Yoshida T, Kuroiwa Y . Induction by bufalin of differentiation of human leukemia cells HL60 U937 and ML1 toward macrophage/monocyte-like cells and its potent synergistic effect on the differentiation of human leukemia cells. Cancer Res 1992; 52: 4634–41.
Krenn L, Kopp B . Bufadienolides from animal and plant sources. Phytochemistry 1998; 48: 1–29.
Song H, Guo T, Bi K, Wang H, Zhang R . Determination of resibufogenin and cinobufagin in heart-protecting musk pill by HPLC. Biomed Chromatogr 2000; 14: 130–2.
Kawazoe N, Watabe M, Masuda Y, Nakaio S, Nakaya K . Tiam1 is involved in the regulation of bufalin-induced apoptosis in human leukemia cells. Oncogene 1999; 18: 2413–21.
Yeh JY, Huang WJ, Kan SF, Wang PS . Effects of bufalin and cinobufagin on the proliferation of androgen dependent and independent prostate cancer cells. Prostate 2003; 54: 112–24.
Paine MF, Khalighi M, Fisher JM, Shen DD, Kunze KL, Marsh CL, et al. Characterization of interintestinal and intraintestinal differences in human CYP3A-dependent metabolism. J Pharmacol Exp Ther 1997; 283: 1552–62.
Tateishi T, Watanabe M, Moriya H, Yamaguchi S, Sato T, Kobayashi S . No ethnic difference between Caucasian and Japanese hepatic samples in the expression frequency of CYP3A5 and CYP3A7 proteins. Biochem Pharmacol 1999; 57: 935–9.
Hebert MF . Contributions of hepatic and intestinal metabolism and P-glycoprotein to cyclosporine and tacrolimus oral drug delivery. Adv Drug Deliver Rev 1997; 27: 201–14.
Hamaoka N, Oda Y, Hase I, Asada A . Cytochrome P4502B6 and 2C9 do not metabolize midazolam: kinetic analysis and inhibition study with monoclonal antibodies. Br J Anaesth 2001; 86: 540–4.
Lin YS, Dowling AL, Quigley SD, Farin FM, Zhang J, Lamba J, et al. Co-regulation of CYP3A4 and CYP3A5 and contribution to hepatic and intestinal midazolam metabolism. Mol Pharmacol 2002; 62: 162–72.
Patki KC, Moltke LL, Greenblatt DJ . In vitro metabolism of midazolam, triazolam, nifedipine, and testosterone by human liver microsomes and recombinant cytochromes P450: role of CYP3A4 and CYP3A5. Drug Metab Dispos 2003; 31: 938–44.
Kim RB, Wandel C, Leake B, Cvetkovic M, Fromm MF, Dempsey PJ, et al. Interrelationship between substrates and inhibitors of human CYP3A and P-glycoprotein. Pharm Res 1999; 16: 408–14.
Acknowledgements
The work was supported by a program for Changjiang Scholars and the Innovative Research Team in University (No PCSIRT), NCET Foundation, NSFC (No 30725045), National 863 Program (No 2006AA02Z338), China Postdoctoral Science Foundation (No 20070410711), “973” program of China (No 2007CB507400), Shanghai Leading Academic Discipline Project (No B906) and in part by the Scientific Foundation of Shanghai, China (No 07DZ19728, 06DZ19717, 06DZ19005).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Li, Hy., Xu, W., Zhang, X. et al. Bufalin inhibits CYP3A4 activity in vitro and in vivo. Acta Pharmacol Sin 30, 646–652 (2009). https://doi.org/10.1038/aps.2009.42
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2009.42
