
Abstract
Comprehensive genetic screening programs have led to the identification of pathogenic methyl-CpG-binding protein 2 (MECP2) mutations in up to 95% of classical Rett syndrome (RTT) patients. This high rate of mutation detection can partly be attributed to specialised techniques that have enabled the detection of large deletions in a substantial fraction of otherwise mutation-negative patients. These cases would normally be missed by the routine PCR-based screening strategies. Here, we have identified large multi-exonic deletions in 12/149 apparently mutation-negative RTT patients using multiplex ligation-dependent probe amplification (MLPA). These deletions were subsequently characterised using real-time quantitative PCR (qPCR) and long-range PCR with the ultimate aim of defining the exact nucleotide positions of the breakpoints and rearrangements. We detected an apparent deletion in one further patient using MLPA; however, this finding was contradicted by subsequent qPCR and long-range PCR results. The patient group includes an affected brother and sister with a large MECP2 deletion also present in their carrier mother. The X chromosome inactivation pattern of all female patients in this study was determined, which, coupled with detailed clinical information, allowed meaningful genotype–phenotype correlations to be drawn. This study reaffirms the view that large MECP2 deletions are an important cause of both classical and atypical RTT syndrome, and cautions that apparent deletions detected using high-throughput diagnostic techniques require further characterisation.
Similar content being viewed by others

Introduction
Rett syndrome (RTT; OMIM no. 312750) is an X-linked neurodevelopmental disorder affecting females almost exclusively. RTT affects approximately 1 in 8300 females before the age of 15 years,1 making it one of the major causes of severe mental retardation in females. RTT is characterised by a distinctive set of clinical features, including a loss of motor skills and communicative abilities, acquired microcephaly and the development of stereotypical hand movements.2 These features typically manifest between 6–18 months of age after a period of apparently normal development.
RTT is caused by loss-of-function mutations in the methyl-CpG-binding protein 2 (MECP2) gene on Xq28;3 over 99% of cases are sporadic and represent de novo mutations.4 Familial cases, although rare, are usually explained by germline mosaicism or skewed X chromosome inactivation (XCI) in the carrier mother. The development of comprehensive diagnostic screening strategies has led to the identification of MECP2 mutations in up to 95% of classical RTT patients worldwide.5 Many groups have reported on the MECP2 mutation spectrum observed in their respective patient cohorts. The eight most common missense and nonsense mutations, all located within exons 3 and 4, constitute approximately two-thirds of all mutations (RettBASE: http://mecp2.chw.edu.au).6 Small deletions (20–500 bp) in the 3′ end of exon 4 make up approximately a further 10% of all mutated alleles.
To account for the small proportion of RTT patients in whom no pathogenic mutations were being identified, some groups postulated that a second locus was involved and searched for new candidate genes associated with RTT.7 Other groups speculated that larger deletions might occur, but would be missed by the routine non-quantitative PCR-based screening strategies because of exclusive amplification of the normal allele.8, 9 Accordingly, several groups have developed techniques to screen apparently mutation-negative RTT patients for the presence of large multi-exonic deletions.10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21 These groups have detected large MECP2 deletions in 56/202 (27.7%) classical RTT patients in whom prior analysis of coding sequences revealed a seemingly wild-type result. These deletions typically affect exons 3 and 4, with many breakpoints falling within a common deletion-prone region (DPR) at the 3′ end of exon 4.15
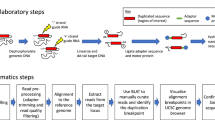
Here, we use a real-time quantitative PCR (qPCR) approach to characterise large MECP2 deletions in 12/149 apparently mutation-negative RTT patients initially detected using multiplex ligation-dependent probe amplification (MLPA). These deletions were then fine-mapped to the nucleotide level using long-range PCR for six patients. The patient group includes six females with classical RTT, five females with atypical RTT and one male with a severe neonatal encephalopathy. In one additional case (Patient 13), MLPA detected a deletion, which was not confirmed by subsequent qPCR and long-range PCR results. We report here a family with an affected brother and sister, both of whom have a large ∼8.5 kb MECP2 double deletion, which is also present in their carrier mother. Subsequent analysis revealed that the mother has highly skewed XCI leading to preferential silencing of the mutant allele, whereas her classical RTT daughter has a random pattern of XCI. This novel family represents not only the first genetically confirmed case of familial RTT in Australia, but also the first male patient worldwide with a large deletion of the MECP2 gene.
Materials and methods
Patients and DNA samples
This study involved 149 patients in whom no pathogenic mutations were identified using routine PCR-based screening of the four MECP2 exons. Of the 149 patients, 48 (including Patients 1–12 in this study) are registered with the Australian Rett Syndrome Database (ARSD).22 As at mid 2007, total ARSD registration includes 312 verified Australian cases, which have been well characterised clinically as classical or atypical RTT and were born in 1976 and subsequently. As has been described previously,1 verification of cases in the ARSD (as either classical or atypical) currently requires the presence of at least six of the eight necessary criteria originally set out by the Rett Syndrome Diagnostic Criteria Work Group23 and modified in 2002,24 provided either criterion five (loss of hand skills) or seven (stereotypic hand movements) is present. A minimum of three primary inclusion criteria from Hagberg's variant delineation model also have to be present.25 Categorisation of cases as classical requires the presence of all the criteria as revised in 2002.24 Clinical information was gathered on patients from the ARSD and through direct contact with clinicians. Where possible, patients were assigned overall clinical severity scores for the Kerr, Percy and Pineda scales, as described previously.22 Genotype–phenotype correlations were made using SPSS v 13.0 software (SPSS Inc., Chicago, IL, USA).
The remaining patients had also been referred to the Department of Molecular Genetics at the Children's Hospital, Westmead, for MECP2 mutation testing, but had not been reported or did not meet the criteria for eligibility to the ARSD. The total patient group includes one male (Patient 8), who is the brother of Patient 9. DNA samples were obtained from all patients with informed consent, as well as the mother of Patients 8 and 9 (the children have different fathers, neither of whom were available for testing). DNA was extracted from peripheral blood leucocytes using the ‘salting out’ method26 and diluted appropriately. Appropriate ethical approval for this study has been obtained from the Ethics Department at the Children's Hospital, Westmead.
Case report: affected RTT family
Patient 9, currently aged 5 years, has classical RTT. Pregnancy and delivery were normal. Developmental delay was first noted at the age of 6 months; by 1 year of age, global developmental regression was evident. Deceleration in head growth was noticed at 1 year of age. Her head circumference is currently below the second percentile. At the most recent assessment, her weight was at the third percentile. She had lost all purposeful hand movements by the age of 18 months, and has since developed a range of stereotypical hand movements such as frequent hand wringing and chest tapping. She has experienced seizures, which are easily controlled with sodium valproate. She has never walked, never developed any meaningful speech, is wheelchair-bound and is dependent for all care. She has frequent bruxism, constipation, scoliosis and irregular breathing patterns. EEG showed intermittent asynchrony between the two hemispheres of the brain with potentially epileptogenic changes, whereas MRI was suggestive of mild generalised atrophy. She had a normal 46XX karyotype, and basic metabolic investigations of blood and urine were normal.
Patient 8, currently aged 18 months, presented with a severe neonatal encephalopathy phenotype. The pregnancy was complicated by PV bleeding, which was originally assumed to have been a spontaneous abortion until pregnancy was ‘re-diagnosed’ at 20 weeks. He was born via caesarean section at 38 weeks for a low-lying placenta. He weighed 2810 g and subsequently spent 3 weeks in a special care nursery due to poor feeding, failure to thrive and apnoea. He has had poor somatic and head growth; head circumference currently 1 cm below the second percentile; weight 1.6 kg below the third percentile and length on the third percentile. He has a scaphocephaly and myopathic-looking facies. He has a prominent rotary nystagmus, which has attenuated with age, and striking bilateral pupillary constriction (miosis). He has periodic breathing while awake and asleep. He has also developed hand mannerisms, with his fists invariably clenched and placed in his mouth or ear, but has no dystonia. He has marked central hypotonia with poor muscle bulk, early flexion contractures at the knees and brisk deep tendon reflexes and a crossed adductor response. He has no obvious visual function, speech or eye contact. MRI shows a structurally normal brain, and basic metabolic tests have all been normal. He developed generalised motor seizures at the age of 18 months. To date, he has not reached any clear developmental milestones, but his condition does not appear to be deteriorating. The mother identified attentional problems at school, but subsequently obtained a diploma in clerical skills. She has longstanding severe anxiety and depression. She also has infrequent hand tremors, but none of the criteria normally associated with RTT seen in her children.
MLPA analysis
All 149 patients who tested negative for MECP2 mutations in coding sequences were analysed using MLPA. MECP2-MLPA was performed using kit P015C (MRC-Holland, Amsterdam, The Netherlands) as described previously.14, 27 This assay covers all four MECP2 exons and the flanking genes IRAK1, L1CAM and SYBL1.
qPCR analysis
To narrow down the deletion breakpoints in each patient, we used real-time qPCR to test the relative copy number of various strategically designed amplicons located along the MECP2 gene. Primers were designed from the genomic clone AF030876 with the help of the Primer3 program28 (primer sequences and annealing sites available upon request). Briefly, our qPCR strategy was based on generating standard curves for each MECP2 amplicon, and also for an autosomal reference gene (GAPDH); these standard curves define the relationship between the input DNA concentration and the Ct value. Copy number standards were produced by amplifying the respective fragment from genomic DNA on a PCR Express thermocycler (Hybaid, Franklin, MA, USA) using Brilliant SYBR Master Mix (Stratagene, La Jolla, CA, USA). Amplicons were gel purified using the QIAquick Gel Extraction Kit (Qiagen, Hilden, Germany) and then serially diluted based on concentration determined by spectrophotometry (Beckman Coulter, Fullerton, CA, USA). All protocols were carried out essentially in accordance with the manufacturer's instructions with some optimisation.
Real-time qPCR was performed using a Rotor-Gene 3000 A thermocycler (Corbett Research, Sydney, Australia). All reactions were conducted in triplicate, with the average of each triplicate group used for quantitative analysis. Product specificity was assessed using melt curve analysis and gel electrophoresis of qPCR products. The MECP2 amplicon of interest and the GAPDH reference amplicon were amplified separately for each patient and also for a normal female control, yielding a copy number for each. The copy number of the MECP2 amplicon was divided by the reference GAPDH amplicon to normalise for slight differences in input DNA concentrations. The normalised copy number for each patient was calibrated to the normal female control included in each run. An RTT deletion-positive female or a normal (hemizygous) male was included in each qPCR run to act as a positive control.
Long-range PCR amplification and sequencing of deletion junctions
When deletion breakpoints had been narrowed down to a sufficiently small region using qPCR, primer sites in the dizygous regions immediately flanking the breakpoints were selected for long-range PCR amplification across the deletion junction. As the precise size of the junction fragment in each patient was unknown, several different PCR conditions were tested and optimised. If the expected fragment size was less than 2 kb, long-range PCR was performed using AmpliTaq Gold (Applied Biosystems, Foster City, CA, USA); the Expand Long Template PCR System (Roche, Mannheim, Germany) was used for larger products. Both protocols were carried out in accordance with the manufacturer's instructions on a PCR Express thermocycler.
When a specific PCR product was generated, it was treated with 1 U shrimp alkaline phosphatase (Promega, Madison, WI, USA) and 5 U exonuclease I (Epicentre, Madison, WI, USA) at 37°C for 30 min, followed by a deactivation step at 80°C for 15 min. Sequencing was carried out on an ABI Prism 3100 Genetic Analyser using BigDye Terminator v 3.1 chemistry (Applied Biosystems). Sequences were aligned with the normal MECP2 genomic sequence (obtained from the genomic clone AF030876) to determine the exact positions of breakpoints and rearrangements; we define nucleotide 1 as the transcriptional start site of MECP2 exon 1. Sequences surrounding the deletion junctions were examined for the presence of repetitive elements and other factors that may shed light upon the mechanisms involved in mutagenesis.
X Chromosome inactivation assay
The XCI status of all 12 female patients, as well as the mother of Patients 8 and 9, was determined by investigation of the methylation status of the highly polymorphic X-linked androgen receptor (AR) locus.29 For each subject, 500 ng of genomic DNA was digested separately with HhaI and McrBC restriction enzymes (New England Biolabs, Beverly, MA, USA) in accordance with the manufacturer's instructions. Enzyme digests were carried out in duplicate to control for incomplete digestion. A ∼280 bp region of the AR locus was PCR amplified from digested and undigested DNA using fluorochrome-labelled primers. Samples were electrophoresed on an ABI Prism 3100 Genetic Analyser and peak areas quantified using GeneScan v 3.7 software (Applied Biosystems). A known highly skewed RTT female and a normal male were included in the analysis to act as controls.
Results
Detection of deletions using MLPA
Of the 149 patients without a detected mutation in MECP2 coding regions, MLPA detected one or more missing exonic probes in 13 patients. For some of the remaining patients, available DNA was of insufficient quality. The same missing probes from exons 3 and 4 were detected in all three available members of the affected family.
Characterisation of deletions in patients using qPCR
In all patients with suspected deletions on the basis of MLPA, qPCR analysis of the respective region yielded results compatible with a deletion, except in the case of Patient 13. In this case, qPCR analysis of the suspected region in exon 4 produced results suggestive of a normal dizygous genotype. In all other patients, the PCR-jumping strategy was successfully applied; relative ratios of 0.35–0.73 were suggestive of a deletion, whereas ratios of 0.82–1.28 were indicative of a normal dizygous copy number for that region. Deletion breakpoints were narrowed down to less than 1-kb regions in all cases except Patients 3 and 12. For these two patients, the upstream breakpoint was localised within intron 2, whereas the downstream breakpoints extended well into the IRAK1 locus beyond the coverage of the AF030876 genomic clone. In each run, relative ratios suggestive of a deletion were reliably obtained for the hemizygous control (data not shown), confirming the accuracy of our qPCR assay. For Patient 8, the only male in the study, simple (non-quantitative) PCR was performed to localise the deletion breakpoints. Subsequent long-range PCR results (see below) made it unnecessary to conduct qPCR on the mother or sister of Patient 8.
Long-range PCR amplification across deletion junctions
Long-range PCR across the breakpoints was used to further characterise the deletions in the 9 patients for whom the approximate positions of the upstream and downstream breakpoints were defined by qPCR. Long-range PCR approaches were successful in five cases (Patients 1, 7, 8, 10 and 11; Figure 1). In many cases, several different forward/reverse primer combinations were tested before success was achieved (data not shown). For Patients 2, 4, 5 and 6, multiple long-range PCR approaches were designed and tested but ultimately failed. In the case of the affected family, long-range PCR was first performed and optimised on the son (Patient 8). The same PCR assay was then used as a diagnostic tool to assess his sister (Patient 9) and mother for the presence of a familial deletion. The same junction fragment was observed in all three family members (Figure 2).

Long-range PCR results. The gel shows PCR products spanning the deletion breakpoints in Patients 1, 7, 8, 10 and 11. Different forward/reverse primer combinations were used to generate these fragments. In lanes 3 and 5, the normal allele has also amplified in the normal female control. L, DNA ladder; P1, Patient 1; N, normal female control DNA. The numbers on the left and right refer to the sizes of the DNA ladder fragments in kb.

Long-range PCR results for the affected family. The gel shows the ∼1.5 kb PCR product spanning the deletion breakpoints in all three family members. The primers used to generate this fragment are normally ∼10 kb apart in a wild-type allele. The mother (M) is a carrier of the ∼8.5 kb deletion, which she has passed on to her affected son (P8) and daughter (P9). Lane 6 (−)=no template control.
Long-range PCR was also attempted for Patient 13, despite the fact that qPCR analysis found no evidence of a deletion. MLPA screening suggested that this patient had a hemizygous copy number for MLPA probe 3026-L2372; this suspected deletion was detected in two independent MLPA runs. This probe is located in the DPR in the 3′ end of exon 4, making it plausible that this patient had a small deletion affecting the MLPA probe binding site. Multiple long-range PCR approaches were implemented, in which forward and reverse primers were chosen on either side of the two neighbouring nondeleted MLPA probe-binding sites. The PCR product obtained from Patient 13 was always the same size as that observed from a normal female control. Subsequent sequencing of an approximately 2 kb fragment spanning the probe-binding site from Patient 13 revealed a wild-type sequence, with no small deletions or polymorphisms that might affect the binding of the MLPA probe in question. Based on these results, we conclude that this patient does not have a deletion, making the MLPA diagnosis a false positive.
Fine mapping of deletions and rearrangements
We succeeded in sequencing the junction fragments obtained from long-range PCR in the five patients mentioned above. The precise nucleotide positions of the breakpoints and rearrangements were identified and confirmed by both forward and reverse sequencing reactions. As most PCR products were too large to sequence using one primer on each strand, new nested sequencing primers were designed and used. The sizes of the amplified junction fragments corresponded to those expected from the sequencing results in all cases. The nature of the deletions and rearrangements in each patient are summarised in Table 1 and Figure 3.

Schematic representation of the locations of the deletions identified. The six deletions at the top (light bars) were successfully fine mapped to the nucleotide level, including the double deletion found in the affected family. The six deletions at the bottom (black bars) show the approximate locations of the deletions in the remaining patients, based on qPCR results to date. Each of these deletions extends into the downstream IRAK1 gene, represented by the arrow. The question marks (?) indicate that the downstream extent of the deletions in Patients 3 and 12 is unknown. The four bars in the MECP2 gene indicate the positions of exons 1, 2, 3 and 4 (from left to right). Diagram is drawn to scale.
For the affected family, the 1.5 kb junction fragments were sequenced in all three family members, revealing the presence of a double deletion involving exons 3 and 4 (Figure 4). The most upstream breakpoint is located in intron 2, with the most downstream breakpoint in the DPR of exon 4. The two deletions are separated by an intact 13-bp section of intron 3. The same four breakpoints were observed in all three cases. Two other patients (1 and 7) had complex insertion/deletion (indel) mutations.

Sequencing results from the son of the affected family (Patient 8). The sequence trace reveals a double deletion of exons 3 and 4, separated by an intact 13 bp section of intron 3 (boxed sequence). The same sequencing results were observed for his sister (Patient 9) and mother (data not shown).
Analysis of junction sequences
In the six rearranged alleles that were fine mapped to the nucleotide level, we looked for repetitive elements surrounding the deletion breakpoints and also for regions of homology between the upstream and downstream wild-type sequences. All six alleles had either the χ sequence or the GCTGG pentanucleotide present in the region surrounding at least one breakpoint. In Patient 11, the upstream breakpoint is positioned within an L2 repeat element (g.61315_61538). The upstream breakpoints in Patients 1 and 7 are located 45 bp apart within the same AluSx sequence in intron 2 (g.63149_63442). This AluSx element includes a 26 bp core Alu sequence, which has been found at the breakpoints of several human gene deletions (see Discussion). The breakpoint in Patient 7 overlaps with this 26 bp sequence.
Patients 1 and 7 have complex indel mutations. Patient 1 has an insertion of 9 bp, which corresponds to the reverse complement of nucleotides g.65828_65836, a region within the deleted fragment. Patient 7 has a more complex indel rearrangement, with an insertion of 140 bp. Two different computer programs were used to characterise the nature of the inserted sequence. BLAST analysis revealed that 31 bp of the insert is a unique sequence which comes from the reverse complement of nucleotides g.67079_67109, a region within the deleted fragment near the downstream breakpoint flanking the DPR. RepeatMasker identified 86 bp of the remaining 109 bp as part of an AluSp sequence. Interestingly, further BLAST analysis of the 86 bp AluSp sequence found a match with 100% homology for a 54 bp segment located on chromosome 3p22. The origin of the other 23 bp segment remains elusive.
XCI results
All 13 patients were heterozygous at the AR locus and were thus informative for the assay. The results for each subject are listed in Table 1, along with a summary of all other results obtained. Interestingly, Patient 9 has random XCI (52:48), whereas her mother is highly skewed (93:7). Patients 1, 7, 10 and 11 also have skewed XCI (defined here as ≥75% activity of one X chromosome).
Genotype–phenotype correlations
All 12 patients with deletions in this study have been clinically diagnosed with RTT with the exception of Patient 8, who has a severe neonatal encephalopathy. Clinical severity scores, for the deletion patients in which they were available, are shown in Table 1. Several statistical comparisons were made. For severity scores and deletion sizes, the assumption of normally distributed populations could not be made, and so nonparametric statistical tests were used. The clinical score for the patients with all of exons 3 and 4 deleted (n=7) was not significantly different from those for patients with some of this region intact (n=2; Mann–Whitney test; Kerr: 20.9 vs 29.5, P=0.057, Percy: 29.3 vs 34.4, P=0.057, Pineda: 18.2 vs 21.9, P=0.188). The patients with deletions extending into IRAK1 (n=6) did not have significantly higher clinical scores than those patients whose deletions were confined to within MECP2 (n=3; Mann–Whitney test; Kerr: 24.5 vs 20.0, P=0.519, Percy: 30.6 vs 24.0, P=0.699, Pineda: 20.5 vs 18.0, P=0.699). Furthermore, the average deletion size for the patients with classical RTT (n=6) was not significantly different from that of the atypical RTT patients (n=5; Mann–Whitney test; 11.9 vs 37.0 kb, P=0.927).
There was no significant correlation between deletion size and clinical score (Kerr: Spearman's rho=0.083, P=0.831). Large deletions were found in 6/23 classical RTT patients (26.1%) and in 5/25 atypical patients (20.0%) in whom no mutations were previously detected; these proportions were not significantly different (χ2 test, df=2, P=0.882). The proportion of females with deletions in this study with skewed XCI (5/13; 38.5%; 95% confidence interval: 13.9–68.4%) was significantly greater than the proportion of skewed females in the general population (∼10% for females <60 years30). Finally, the average severity scores for the patients in this study (n=9) were compared with the average scores for all mutation-positive patients in the ARSD without large deletions for whom clinical scores were available (n=151). The patients in this study had higher average scores for all three clinical scales, although these differences were not significant (T test, df=158; Kerr: 23.4 vs. 20.8, P=0.211, Percy: 28.3 vs 24.4, P=0.067, Pineda: 18.8 vs 16.1, P=0.077).
Discussion
In this study, we have screened a cohort of apparently mutation-negative RTT patients for large MECP2 deletions using MLPA. One or more missing exonic probes were detected in 13/149 patients; subsequent qPCR analysis has confirmed the presence of large deletions in 12 of these patients. In these patients, the deletion breakpoints were further characterised using qPCR and long-range PCR with the ultimate aim of defining the precise endpoints at the nucleotide level. The latter was achieved for 6 of these 12 patients.
Previous studies have reported deletions in 71/450 (15.8%) RTT patients in whom a wild-type sequence was previously found for all exons, including both classical and atypical cases. The lower proportion in this study (8.1%; 12/149) is probably due to the inclusion of a wider spectrum of patients. Amongst our cohort of patients screened in the diagnostic laboratory, many were selected only on the basis of referral for MECP2 analysis. Hence, not all patients from this cohort have been clinically diagnosed with RTT, and re-evaluation of their clinical diagnoses is being considered. On the other hand, if we consider the proportion in those cases confirmed by the ARSD, it is considerably higher at 25% (12/48).
Before this study, there were only five reported atypical RTT patients with large MECP2 deletions.16, 20 This study has identified a further five atypical RTT cases with large deletions, along with a male patient with a severe neonatal encephalopathy. Our rate of detection of large deletions in classical and atypical RTT patients previously thought to be mutation negative were not significantly different, similar to previous findings.15 These findings would support the view that the relative lack of large deletions found in atypical RTT patients could merely reflect a patient selection bias in that classic RTT patients constitute the most intensively studied group in terms of MECP2 gene analysis.20 Our study reaffirms the view that large MECP2 deletions are an important and not infrequent cause of atypical RTT.
All the deletions reported in this study involve exon 3 and/or 4, and in most cases both. This is in accordance with previous findings, although a small number of deletions have been reported affecting exons 1 and 2.14, 18, 20 We have identified one additional patient with a large deletion of MECP2 exons 1 and 2 using MLPA (unpublished data), although this is a rare phenomenon in our experience. Whereas it has been suggested that this could be due to an ascertainment bias introduced by the genomic location of amplicons used,15 this can be excluded in our case, as the MLPA assay includes amplicons covering all four MECP2 exons and also three neighbouring genes. We were unable to further characterise the deletion in this patient using our qPCR approach due to an insufficiency of DNA, hence this patient was not included in the present study.
For the five rearranged alleles which we successfully fine mapped to the nucleotide level, only one had a breakpoint in the DPR, as defined by Laccone et al.15 One other patient had a potential breakpoint in the DPR (Patient 2). Five out of our 12 deletion patients had a breakpoint in the immediate vicinity of the DPR. Combining our findings with the three previous studies which have defined the precise endpoints of large MECP2 deletions,13, 15, 18 12/21 (57.1%) rearranged alleles have a breakpoint in the DPR. It is quite remarkable that the breakpoints of these large multi-kilobase deletions are restricted to a common ∼150-bp region; interestingly, this region is also the hotspot for the smaller deletions (20–500 bp) confined within exon 4. Several explanations have been proposed to account for the intrinsic instability of this region; these include the presence of direct and inverted repeats,8 the abundance of polypurine residues in the antisense strand31 and the presence of a so-called χ sequence,15 which has been found to be highly recombinogenic in the Escherichia coli genome.32 However, the χ sequence is found in an additional four locations in the AF030876 genomic clone, none of which have been reported to be recombination hotspots. It is likely that the mutagenic nature of the DPR is caused by synergistic interactions between several different recombinogenic factors.
It has been proposed that large MECP2 deletions are often caused by potent mutagenic interactions between χ-like sequences and Alu repeats in intron 2.15 The rearrangements in Patients 1 and 7 in the present study lend support to this hypothesis. The upstream breakpoint in these patients is positioned within the same AluSx element in intron 2, whereas an internal element of the χ sequence (GCTGG) is present near the downstream breakpoint. Interestingly, a 26 bp core Alu sequence which has been found at the breakpoints of many pathogenic human gene deletions33 is also present at the upstream breakpoint in these two patients. These are the same two patients in whom complex indel mutations were identified, suggesting that they may be the result of Alu-mediated indel events. Interestingly, the GCTGG pentanucleotide is found in both the χ sequence and in the 26 bp core Alu sequence.15 Analysis of the AF030876 genomic clone with the RepeatMasker program (http://www.repeatmasker.org) revealed that 22.8% of its sequence consists of Alu elements. Accordingly, one would expect 22.8% of deletion breakpoints to fall within an Alu element purely by chance. Taking our findings together with the three above mentioned studies, 11/21 (52.4%; 95% confidence interval: 29.8–74.3%) rearranged alleles have had at least one breakpoint within an Alu sequence. This observed proportion is significantly greater than what would be expected due to chance alone, suggesting that Alu repeats do indeed play a role in mutagenesis, probably by facilitating illegitimate recombination.
As all of the deletions in this study affect exon 3 and/or 4, they affect crucial regions of the MeCP2 protein and can be assumed to be pathogenic. The methyl-CpG-binding domain (MBD) spans exons 3 and 4, whereas the transcription repression domain (TRD) is located within exon 4. These two functional domains are completely deleted in 10/12 patients with deletions in this study. For these patients, it is highly unlikely that the protein produced from the mutant allele (if any) would have residual function; the putative proteins would be unable to enter the nucleus, let alone bind target genes or repress transcription. It might be expected that the patients with the MBD and TRD partially or wholly intact would present with a relatively mild phenotype. However, our patients with all of exons 3 and 4 deleted did not have significantly different phenotype scores from the patients with some of this region intact. The deletion in Patient 2 leaves exon 3 intact, with the upstream breakpoint located in exon 4. Surprisingly, she has the most severe phenotype of all the patients in the group for whom clinical scores were available; she also has random XCI in blood leucocytes. In her case, however, the entire 3′ UTR is deleted, along with the first ∼2 kb of the IRAK1 gene, which probably adds to the severity of her phenotype. It should be noted that she was also the eldest of the deletion patients and therefore her clinical scores are likely to be associated with some age-related changes.
In Patient 10, both the MBD and the TRD are left completely intact. The last 229 bp of exon 4 are deleted, including the stop codon, along with a further 2571 bp of the 3′ UTR. This would, theoretically, produce a MeCP2 protein truncated after residue 409. This leads to the loss of almost half of the recently characterised WW domain. Based on functional studies conducted by others, this would lead to severely reduced, if not totally abolished, binding activity of this domain.34 Unfortunately, total clinical severity scores were not available for this patient, although she did appear to have a relatively mild phenotype based on the limited scores that were available for individual scoring criteria. Notably, she has not experienced seizures, which is unusual amongst our group of patients. She has been diagnosed with atypical RTT. Whether large MECP2 deletions invariably result in a total loss of protein function is yet to be determined. This could be further explored through transcript analysis or, ideally, functional proteomic studies.
We have attempted to draw genotype–phenotype correlations in our large deletion patient cohort. One group has recently proposed that deletions extending into the downstream IRAK1 gene may make the phenotype more severe.20 Three other groups have also reported such deletions,15, 18, 21 although none of the affected patients have had additional clinical features. In our study, six patients have deletions extending into the IRAK1 gene; the average severity score for these patients was higher than in the other six patients whose deletions were located wholly within MECP2. This was observed for all three clinical scores, although the differences were not statistically significant. In Patients 2, 4, 5 and 6, the downstream breakpoints were identified in the proximal part of IRAK1, and so any disruption to IRAK1 would be minimal. In Patients 3 and 12, however, it appears that much more of the IRAK1 gene is deleted. These two patients have random XCI in blood leucocytes. Patient 12, now deceased, had a particularly severe phenotype that was not fully reflected in her clinical scores at 8 years of age. It is believed that her death at the age of 9 years was associated with complications related to her condition, although the nature of her additional clinical features is not known in detail. Patient 3 has a fairly moderate phenotype despite the involvement of the IRAK1 gene. IRAK1 is essential for the regulation of innate immunity,35 and has been associated with atherosclerosis36 and myocardial contractile dysfunction.37 Due to the involvement of the IRAK1 locus in cardiac function, we suggest that patients with deletions extending into IRAK1 should be considered for clinical assessment of underlying cardiovascular abnormalities.
The average deletion size in atypical RTT patients was larger than that in classical cases, but this difference was not significant. This is not surprising as atypical patients can be either more or less severely affected than classical patients; even if larger deletions did have a tendency to lead to more severe phenotypes this would not necessarily lead to either the classical or atypical phenotype predominating. There did appear to be a positive trend between deletion size and clinical severity, although no significant relationship was observed for any of the three severity scores. We acknowledge that this may well be due to poor statistical power associated with our sample size.
Archer et al20 noted that their deletion group were indistinguishable from other mutation-positive RTT patients in terms of average clinical severity. The patients in our study had higher clinical severity scores, on average, than all the mutation-positive patients without large deletions in the ARSD. This was true for all three severity scores, although the differences were not significant at the 5% level. Whereas the numbers in this study are too small to draw definite conclusions, it seems plausible that RTT patients with large deletions are more severely affected than their mutation-positive counterparts without large deletions. It is quite likely that large multi-exonic deletions result in a greater loss of protein function than would be expected for other common types of mutations such as missense, nonsense and frameshift mutations. The only type of mutation that would conceivably be more severe than a large deletion would be a nonsense mutation that leads to premature truncation of the protein before exon 3.
In this study, we have reported a family in which multiple members harbour a large MECP2 deletion. The carrier mother has passed on the mutation to her affected son and daughter, as demonstrated by our long-range PCR results in all three family members. To our knowledge, this is the first male patient reported worldwide with a large MECP2 deletion, although recently some male subjects have been reported with duplications of the Xq28 chromosomal band.38, 39, 40, 41, 42, 43 He is only the 16th male in which a pathogenic MECP2 mutation associated with early post-natal encephalopathy has been found.44 As this is a severe mutation, and the possibility of mosaicism can be excluded due to the familial inheritance of the allele, this case provides strong evidence that an absence of functional MeCP2 is compatible with human life. Although this individual has a severe neonatal encephalopathy, his condition does not appear to be deteriorating at present. The finding of a mutation present in all three family members indicated that the mother either has highly skewed XCI or is a germline mosaic; one of these has been the case in other instances of familial RTT.45, 46, 47, 48, 49, 50, 51, 52 We found a highly skewed pattern of XCI in the mother's blood leucocytes, whereas her daughter has random XCI. By comparing the sizes of the AR alleles in the mother and daughter, we were able to establish that the mutant allele is preferentially silenced in the mother. This family illustrates the role that XCI plays in modulating the RTT phenotype. We have noticed some evidence of a mild learning disability and fine hand tremor in the mother, although these abnormalities were only detected in retrospect after a familial mutation was identified. A family with two RTT daughters with large deletions of exons 3 and 4 has recently been reported (F Ariani et al, personal communication), but due to an absence of the mutation in either parent, it was hypothesised that one of the parents is a germline mosaic.
In the past, many laboratories have overlooked large heterozygous deletions due to their reliance on PCR-based screening methods. This study, combined with the findings of previous reports, highlights the importance of screening both classical and atypical RTT patients for large MECP2 deletions. Large deletions of exon 3 and/or 4 accounted for 12/211 (5.7%) of all pathogenic mutations in the ARSD patient cohort. This makes large deletions as a group one of the most frequent MECP2 mutations in the cohort, a phenomenon also reported by Archer et al.20 Using MLPA, we identified deletions in 13/149 apparently mutation-negative patients, although in one patient (Patient 13), subsequent qPCR and long-range PCR analysis found no evidence of a deletion. As the missing exon 4 probe was detected in two independent MLPA runs, it is likely to be a reproducible MLPA artefact. We cannot, however, rule out the possibility that this patient harbours a complex underlying mutation that destroys the MLPA probe binding site leading to exclusive amplification of the normal allele during long-range PCR. We have recently come across a similar case in which an insertion of genetic material disturbed an MLPA probe-binding site; in this latter case, however, we succeeded in amplifying a larger product from the mutant allele (unpublished data). Our results in the case of Patient 13 show that, if a similar phenomenon has occurred, the mutant allele is beyond the scope of normal PCR amplification. We are considering screening this patient with a third independent method such as Southern blotting to resolve the issue. Our findings in this patient demonstrate the need to further characterise apparent deletions detected by high-throughput diagnostic methods such as MLPA.
References
Laurvick CL, de Klerk N, Bower C et al: Rett syndrome in Australia: a review of the epidemiology. J Pediatr 2006; 148: 347–352.
Weaving LS, Ellaway CJ, Gecz J, Christodoulou J : Rett syndrome: clinical review and genetic update. J Med Genet 2005; 42: 1–7.
Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY : Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 1999; 23: 185–188.
Trappe R, Laccone F, Cobilanschi J et al: MECP2 mutations in sporadic cases of Rett syndrome are almost exclusively of paternal origin. Am J Hum Genet 2001; 68: 1093–1101.
Amir R, Sutton V, Van den Veyver I : Newborn screening and prenatal diagnosis for Rett syndrome: implications for therapy. J Child Neurol 2005; 20: 779–783.
Christodoulou J, Grimm A, Maher T, Bennetts B : RettBASE: the IRSA MECP2 variation database—a new mutation database in evolution. Hum Mutat 2003; 21: 466–472.
Renieri A, Meloni I, Longo I et al: Rett syndrome: the complex nature of a monogenic disease. J Mol Med 2003; 81: 346–354.
De Bona C, Zappella M, Hayek G et al: Preserved speech variant is allelic of classic Rett syndrome. Eur J Hum Genet 2000; 8: 325–330.
Laccone F, Huppke P, Hanefeld F, Meins M : Mutation spectrum in patients with Rett syndrome in the German population: evidence of hot spot regions. Hum Mutat 2001; 17: 183–190.
Bourdon V, Philippe C, Grandemenge A, Reichwald K, Jonveaux P : Deletion screening by fluorescence in situ hybridization in Rett syndrome patients. Ann Genet 2001; 44: 191–194.
Bourdon V, Philippe C, Labrune O, Amsallem D, Arnould C, Jonveaux P : A detailed analysis of the MECP2 gene: prevalence of recurrent mutations and gross DNA rearrangements in Rett syndrome patients. Hum Genet 2001; 108: 43–50.
Yaron Y, Ben Zeev B, Shomrat R, Bercovich D, Naiman T, Orr-Urtreger A : MECP2 mutations in Israel: implications for molecular analysis, genetic counseling, and prenatal diagnosis in Rett syndrome. Hum Mutat 2002; 20: 323–324.
Schollen E, Smeets E, Deflem E, Fryns J, Matthijs G : Gross rearrangements in the MECP2 gene in three patients with Rett syndrome: implications for routine diagnosis of Rett syndrome. Hum Mutat 2003; 22: 116–120.
Erlandson A, Samuelsson L, Hagberg B, Kyllerman M, Vujic M, Wahlstrom J : Multiplex ligation-dependent probe amplification (MLPA) detects large deletions in the MECP2 gene of Swedish Rett syndrome patients. Genet Test 2003; 7: 329–332.
Laccone F, Jünemann I, Whatley S et al: Large deletions of the MECP2 gene detected by gene dosage analysis in patients with Rett syndrome. Hum Mutat 2004; 23: 234–244.
Ariani F, Mari F, Pescucci C et al: Real-time quantitative PCR as a routine method for screening large rearrangements in Rett syndrome: report of one case of MECP2 deletion and one case of MECP2 duplication. Hum Mutat 2004; 24: 172–177.
Huppke P, Ohlenbusch A, Brendel C, Laccone F, Gärtner J : Mutation analysis of the HDAC 1, 2, 8 and CDKL5 genes in Rett syndrome patients without mutations in MECP2. Am J Med Genet 2005; 137A: 136–138.
Ravn K, Nielsen JB, Skjeldal OH, Kerr A, Hulten M, Schwartz M : Large genomic rearrangements in MECP2. Hum Mutat 2005; 25: 324.
Shi J, Shibayama A, Liu Q et al: Detection of heterozygous deletions and duplications in the MECP2 gene in Rett syndrome by Robust Dosage PCR (RD-PCR). Hum Mutat 2005; 25: 505.
Archer HL, Whatley SD, Evans JC et al: Gross rearrangements of the MECP2 gene are found in both classical and atypical Rett syndrome patients. J Med Genet 2006; 43: 451–456.
Pan H, Li MR, Nelson P, Bao XH, Wu XR, Yu S : Large deletions of the MECP2 gene in Chinese patients with classical Rett syndrome. Clin Genet 2006; 70: 418–419.
Colvin L, Fyfe S, Leonard S et al: Describing the phenotype in Rett syndrome using a population database. Arch Dis Child 2003; 88: 38–43.
The Rett Syndrome Diagnostic Criteria Work Group: Diagnostic criteria for Rett syndrome. Ann Neurol 1988; 23: 425–428.
Hagberg B, Hanefeld F, Percy A, Skjeldal O : An update on clinically applicable diagnostic criteria in Rett syndrome. Comments to Rett Syndrome Clinical Criteria Consensus Panel Satellite to European Paediatric Neurology Society Meeting, Baden Baden, Germany, 11 September 2001. Eur J Paediatr Neurol 2002; 6: 293–297.
Hagberg B : Clinical delineation of Rett syndrome variants. Neuropediatrics 1995; 26: 62.
Miller S, Dykes D, Polesky H : A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 1988; 16: 1215.
Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G : Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res 2002; 30: e57.
Rozen S, Skaletsky H : Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 2000; 132: 365–386.
Allen R, Zoghbi H, Moseley A, Rosenblatt H, Belmont J : Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am J Hum Genet 1992; 51: 1229–1239.
Busque L, Mio R, Mattioli J et al: Nonrandom X-inactivation patterns in normal females: lyonization ratios vary with age. Blood 1996; 88: 59–65.
Lebo RV, Ikuta T, Milunsky JM, Milunsky A : Rett syndrome from quintuple and triple deletions within the MECP2 deletion hotspot region. Clin Genet 2001; 59: 406–417.
Stahl F, Stahl M : Recombination pathway specificity of Chi. Genetics 1977; 86: 715–725.
Rudiger N, Gregersen N, Kielland-Brandt M : One short well conserved region of Alu-sequences is involved in human gene rearrangements and has homology with prokaryotic chi. Nucleic Acids Res 1995; 23: 256–260.
Buschdorf J, Stratling W : A WW domain binding region in methyl-CpG-binding protein MeCP2: impact on Rett syndrome. J Mol Med 2004; 82: 135–143.
Su K, Richter K, Zhang C, Gu Q, Li L : Differential regulation of interleukin-1 receptor associated kinase 1 (IRAK1) splice variants. Mol Immunol 2007; 44: 900–905.
Huang Y, Li T, Sane DC, Li L : IRAK1 serves as a novel regulator essential for lipopolysaccharide-induced interleukin-10 gene expression. J Biol Chem 2004; 279: 51697–51703.
Thomas JA, Haudek SB, Koroglu T et al: IRAK1 deletion disrupts cardiac Toll/IL-1 signaling and protects against contractile dysfunction. Am J Physiol Heart Circ Physiol 2003; 285: 597–606.
Friez MJ, Jones JR, Clarkson K et al: Recurrent infections, hypotonia, and mental retardation caused by duplication of MECP2 and adjacent region in Xq28. Pediatrics 2006; 118: e1687–e1695.
Lugtenberg D, de Brouwer APM, Kleefstra T et al: Chromosomal copy number changes in patients with non-syndromic X linked mental retardation detected by array CGH. J Med Genet 2006; 43: 362–370.
Sanlaville D, Prieur M, de Blois M-C et al: Functional disomy of the Xq28 chromosome region. Eur J Hum Genet 2005; 13: 579–585.
Meins M, Lehmann J, Gerresheim F et al: Submicroscopic duplication in Xq28 causes increased expression of the MECP2 gene in a boy with severe mental retardation and features of Rett syndrome. J Med Genet 2005; 42: e12.
del Gaudio D, Fang P, Scaglia F et al: Increased MECP2 gene copy number as the result of genomic duplication in neurodevelopmentally delayed males. Genet Med 2006; 8: 784–792.
Van Esch H, Bauters M, Ignatius J et al: Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am J Hum Genet 2005; 77: 442–453.
Kankirawatana P, Leonard H, Ellaway C et al: Early progressive encephalopathy in boys and MECP2 mutations. Neurology 2006; 67: 164–166.
Sirianni N, Naidu S, Pereira J, Pillotto R, Hoffman E : Rett syndrome: confirmation of X-linked dominant inheritance, and localization of the gene to Xq28. Am J Hum Genet 1998; 63: 1552–1558.
Wan M, Lee S, Zhang X et al: Rett syndrome and beyond: recurrent spontaneous and familial MECP2 mutations at CpG hotspots. Am J Hum Genet 1999; 65: 1520–1529.
Bienvenu T, Carrie A, de Roux N et al: MECP2 mutations account for most cases of typical forms of Rett syndrome. Hum Mol Genet 2000; 9: 1377–1384.
Villard L, Kpebe A, Cardoso C, Chelly J, Tardieu M, Fontes M : Two affected boys in a Rett syndrome family: clinical and molecular findings. Neurology 2000; 55: 1188–1193.
Villard L, Levy N, Xiang F et al: Segregation of a totally skewed pattern of X chromosome inactivation in four familial cases of Rett syndrome without MECP2 mutation: implications for the disease. J Med Genet 2001; 38: 435–442.
Gill H, Cheadle JP, Maynard J et al: Mutation analysis in the MECP2 gene and genetic counselling for Rett syndrome. J Med Genet 2003; 40: 380–384.
Evans JC, Archer HL, Whatley SD, Clarke A : Germline mosaicism for a MECP2 mutation in a man with two Rett daughters. Clin Genet 2006; 70: 336–338.
Dayer A, Bottani A, Bouchardy I et al: MECP2 mutant allele in a boy with Rett syndrome and his unaffected heterozygous mother. Brain Dev 2007; 29: 47–50.
Acknowledgements
We thank Julianne Jackson and Gemma Jenkins for screening diagnostic patients for MECP2 mutations before MLPA analysis. HL is funded by National Health and Medical Research Council (NHMRC) program Grant 353514. JC is funded by NHMRC project Grants 346603 and 457238. We also acknowledge the funding of Australian RTT research by the National Institutes of Health (1 R01 HD43100-01A1) and the NHMRC under project Grant 303189. Finally, we express our gratitude to the Rett Syndrome Australian Research Fund, the Rotary Club of Narellan, and the Country Women's Association of NSW for financial support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hardwick, S., Reuter, K., Williamson, S. et al. Delineation of large deletions of the MECP2 gene in Rett syndrome patients, including a familial case with a male proband. Eur J Hum Genet 15, 1218–1229 (2007). https://doi.org/10.1038/sj.ejhg.5201911
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201911
Keywords
This article is cited by
-
Ten novel insertion/deletion variants in MECP2 identified in Japanese patients with Rett syndrome
Human Genome Variation (2019)
-
Molecular Testing of MECP2 Gene in Rett Syndrome Phenotypes in Indian Girls
Indian Pediatrics (2018)
-
Clinical and biological progress over 50 years in Rett syndrome
Nature Reviews Neurology (2017)
-
De novo missense mutations in the NAA10 gene cause severe non-syndromic developmental delay in males and females
European Journal of Human Genetics (2015)
-
Custom oligonucleotide array-based CGH: a reliable diagnostic tool for detection of exonic copy-number changes in multiple targeted genes
European Journal of Human Genetics (2013)
