
Abstract
What particular mechanical properties can be expected for materials composed of interlocked backbones has been a long-standing issue in materials science since the first reports on polycatenane and polyrotaxane in the 1970s1,2,3. Here we report a three-dimensional porous metal–organic crystal, which is exceptional in that its warps and wefts are connected only by catenation. This porous crystal is composed of a tetragonal lattice and dynamically changes its geometry upon guest molecule release, uptake and exchange, and also upon temperature variation even in a low temperature range. We indented4 the crystal along its a/b axes and obtained the Young’s moduli of 1.77 ± 0.16 GPa in N,N-dimethylformamide and 1.63 ± 0.13 GPa in tetrahydrofuran, which are the lowest among those reported so far for porous metal–organic crystals5. To our surprise, hydrostatic compression showed that this elastic porous crystal was the most deformable along its c axis, where 5% contraction occurred without structural deterioration upon compression up to 0.88 GPa. The crystal structure obtained at 0.46 GPa showed that the catenated macrocycles move translationally upon contraction. We anticipate our mechanically interlocked molecule-based design to be a starting point for the development of porous materials with exotic mechanical properties. For example, squeezable porous crystals that may address an essential difficulty in realizing both high abilities of guest uptake and release are on the horizon.
Similar content being viewed by others
Main
A variety of mechanically interlocked structures have been found in nature6, of which the geometries and large potential to exert unique functions are of major scientific interest. Historically, the concept of ‘chemical topology’ was introduced for the first time in 1961 and afforded a new challenge in chemistry7. Unlike covalently constructed molecules, mechanically interlocked molecules (MIMs) such as catenanes8,9 and rotaxanes10,11 bear spatially restricted freedom of motion12,13, and a wide variety of synthetic molecular machines and switches based on MIMs have been developed so far14,15. Meanwhile, since the first success in the synthesis of polycatenane and polyrotaxane in the 1970s1,2,3, such mechanically interlocked polymers have attracted particular attention16,17,18,19,20,21 as their densely populated mechanical bonds are expected to move coherently along the main chains, providing anomalous mechanical properties22,23 (Supplementary Table 1). Although peculiar mechanical properties are yet to be reported, precision synthesis of poly[n]catenanes, whose chains are composed solely of catenanes, has been impressive19. We envisaged that if the backbone of a metal–organic crystal were composed of a catenane structure, such a porous crystal might exhibit rubber-like elastic properties. This idea may address the dilemma of porous materials: the higher the guest uptake ability, the less easily guest release occurs. Although most practically useful porous materials are mechanically robust24,25, elastic porous materials, if available, could be mechanically squeezable for the forcing of guest molecules out of the pores. Several different metal–organic crystals26,27 carrying MIMs as backbone pendants28,29,30,31,32,33,34,35 have been reported, exploring their rotary32,33,35 and shuttling34 motions in the solid state. However, as for metal–organic crystals composed of MIM-based backbones, only a few examples have been reported without exploration of their mechanical properties36,37,38,39.
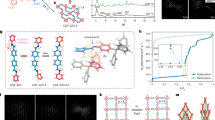
Here, we report a porous metal–organic crystal, [2]catenane-based metal–organic framework (CTNMOF), that is exceptional in that all of its warps and wefts are three-dimensionally (3D) connected only by catenation (Fig. 1b), and it can reversibly deform its crystalline framework in response to an externally applied pressure. The crystal adopts a thin rectangular shape, which technically allows for compression only along the crystallographic a/b axes (crystallographically equivalent to each other), producing Young’s moduli of 1.77 ± 0.16 GPa in N,N-dimethylformamide (DMF) and 1.63 ± 0.13 GPa in tetrahydrofuran (THF). These values are among the smallest of the metal–organic crystals so far reported5, whereas the crystal, when placed under a high hydrostatic pressure, predominantly contracts along the crystallographic c axis. For the synthesis of CTNMOF, we designed a [2]catenane-based40 ligand, H4CTNL, (Fig. 1a, Supplementary Figs. 9–15 and 18), where each of its catenated benzylamide macrocycles carries two carboxylic acid groups, which have the ability to share a metal ion with the adjacent catenane units by coordination. In its crystalline state, these catenated macrocycles are geometrically fixed by an inter-component hydrogen-bonding interaction and adopt either a chair or boat conformation (Supplementary Fig. 19, Supplementary Table 3). By contrast, when Me4CTNL (Supplementary Figs. 4–7, 17, 20, Supplementary Table 4), a methyl ester derivative of H4CTNL, was dissolved in DMF-d7, these hydrogen bonds are attenuated, granting the catenated macrocycles a certain conformational freedom from each other (Supplementary Figs. 2 and 3). When a mixture of H4CTNL and cobalt(II) nitrate hexahydrate (1:8 in mol) in DMF was heated at 90 °C for 18 h, needle-like dark-green crystals of CTNMOF formed (Supplementary Fig. 1). Single-crystal X-ray diffraction (SCXRD) analysis (Supplementary Table 5) at 26 °C revealed that this crystalline material belongs to the chiral space group of P4122 with a composition of [Co2(CTNL)•7H2O•4DMF]n, indicating the occurrence of mirror symmetry breaking in the crystallization process. In regard to this observation, the CTNL units in CTNMOF adopt a chiral topology and are helically arranged along the four-fold screw axis, that is, a axis (Supplementary Fig. 24; left handed, M helicity). As shown in Supplementary Fig. 21e, f, the catenated macrocycles in CTNL form two one-dimensional (1D) metal–organic chains running along the crystallographic a/b axes that are equivalent to each other and correspond to the warps and wefts of the 3D framework of CTNMOF. Consequently, CTNL is located at all of the intersections between the warps and wefts and mechanically interlocks these 1D metal–organic chains (Fig. 1d, Supplementary Fig. 21g, h). Unlike the catenated macrocycles in the H4CTNL crystal (Supplementary Fig. 19), those in CTNMOF all adopt a boat conformation, where two pairs of intramolecular hydrogen bonds are formed between the benzylamide groups. As a result, the crystal composed of the mechanically interlocked warps and wefts carried 3D-interconnected channels (Supplementary Fig. 25) with cross-sectional areas along the c and a/b axes of 6.2 × 6.2 Å2 (Supplementary Fig. 21c) and 4.5 × 4.0 Å2 (Fig. 1c), respectively. By using PLATON41, the solvent-accessible void volume of CTNMOF was calculated to be 59% of the unit cell volume. Accordingly, CTNMOF was able to accommodate gaseous molecules into its crystalline channels. For example, its CO2 adsorption isotherm at −78 °C (Supplementary Fig. 41) showed the presence of a permanent microporous structure with a CO2 sorption capacity of 65 ml STP g−1 at P/P0 = 0.98.

a, Chemical structure of the [2]catenane-based ligand H4CTNL. b, Schematics of CTNMOF, whose warps and wefts are mechanically interlocked by catenation, viewed along the b axis. c, Space-filling representations of CTNMOF viewed along the b axis. d, Wireframe representations of CTNMOF viewed along the b axis. A pair of orthogonally arranged 1D metal–organic chains and a pair of catenated macrocycles of CTNL in CTNMOF are highlighted, where dashed green lines represent hydrogen bonds. Except for those involved in the hydrogen bonds in d, axial ligands of the Co2+-paddle-wheel units, solvent molecules and hydrogen atoms are omitted for clarity.
Crystalline CTNMOF is structurally dynamic42 and undergoes geometrical changes upon guest release, uptake and exchange. As-synthesized CTNMOF in DMF included DMF molecules as guests in its interconnected channels. As observed by thermogravimetric analysis (Supplementary Fig. 39), CTNMOF lost its included DMF molecules upon heating to 100 °C. Notably, guest-free CTNMOF thus obtained showed a high thermal stability and survived up to a temperature of nearly 360 °C without decomposition. The powder X-ray diffraction (PXRD) pattern of CTNMOF soaked in DMF (Fig. 2a, top) agreed well with the simulated pattern (Fig. 2a, bottom) derived from its SCXRD structure (Fig. 1), indicating that CTNMOF has a high phase purity. When the included guest was removed from CTNMOF, its PXRD pattern became relatively featureless (Fig. 2b). However, when guest-free CTNMOF thus obtained was soaked in DMF at 25 °C for 24 h, this solid recovered its original PXRD pattern (Fig. 2c). We also found that the crystal structure of CTNMOF is sensitive to the type of included guest. For example, when DMF-including CTNMOF (Fig. 2a) was soaked in acetonitrile (MeCN) at 25 °C for 1 h, its PXRD pattern changed to a different one, as shown in Fig. 2d. However, when MeCN-soaked CTNMOF was immersed in DMF at 25 °C for 1 h, its PXRD pattern returned to that of DMF-including CTNMOF (Fig. 2e). Meanwhile, when ethanol was used instead of MeCN for soaking DMF-including CTNMOF at 25 °C for 30 min, a single-crystal-to-single-crystal transition took place, where the space group of CTNMOF was transformed from P4122 to P41, as confirmed by SCXRD at −180 °C, as a result of the guest exchange from DMF to ethanol (Fig. 3f, Supplementary Table 11). These observations allow us to conclude that CTNMOF dynamically changes its geometrical feature upon guest release, uptake and exchange while maintaining its porous structure.

a, PXRD pattern of CTNMOF soaked in DMF (top) and its simulated pattern (bottom). b, PXRD pattern of guest-free CTNMOF after being heated at 80 °C for 18 h under a reduced pressure. c, PXRD pattern of heated CTNMOF resoaked in DMF at 25 °C for 24 h. d, PXRD pattern of CTNMOF soaked in MeCN at 25 °C for 1 h. e, PXRD pattern of DMF-soaked CTNMOF immersed in DMF at 25 °C for 1 h.

a–e, Crystal structures of CTNMOF in DMF viewed along the c axis with space groups of P4122 (a), C2221 (b), P41212 (c), P4122 (d) and P212121 (e). f, Crystal structures of CTNMOF in ethanol viewed along the c axis with the space group of P41. The axial ligands of the Co2+-paddle-wheel units, solvent molecules and hydrogen atoms are omitted for clarity. Because the above crystal structures are analogous to each other, the energy barriers required for their transitions are probably small, so that they can be smoothly interconverted without any structural deterioration.
CTNMOF also displayed a thermally adaptive feature. In a set of variable-temperature SCXRD measurements, we collected the diffraction data for a single crystal of DMF-including CTNMOF, upon sequential temperature changes from 26 °C (Figs. 1 and 3a) to −180 °C, −130 °C, −80 °C, −58 °C and finally to −52 °C (Fig. 3b–e, Supplementary Table 12). Upon heating from −180 °C (Fig. 3b, Supplementary Table 6) to −130 °C (Supplementary Table 7), the space group of CTNMOF remained unchanged as C2221. Upon subsequent heating from −130 °C to −80 °C (Fig. 3c, Supplementary Table 8) and then to −58 °C (Fig. 3d, Supplementary Table 9), the space group adaptively changed from C2221 to P41212 and then to P4122, respectively. The space group at −58 °C was the same as that initially observed for DMF-including CTNMOF at 26 °C (Figs. 1 and 3a), which is characterized by the most symmetric polymorph among those observed for CTNMOF, with only one macrocycle from CTNL and one Co2+ ion in the asymmetric unit (Supplementary Fig. 23c). However, when CTNMOF was heated from −58 °C to −52 °C and kept at the same temperature for 24 h, the crystal changed its space group to P212121 (Fig. 3e, Supplementary Table 10), which is the least symmetric geometry among those observed for CTNMOF, with four macrocycles (two CTNL) and four Co2+ ions in the asymmetric unit (Supplementary Fig. 23d). Such a temperature-adaptive structural variation, one of the most characteristic features of CTNMOF due to its 3D catenated backbone, was clearly confirmed from the change in cross-sectional shape of the largest channel running along the c axis (Fig. 3): from a square at 26 °C to a rectangle at −180 °C, a rhombus at −80 °C, a square at −58 °C and then a parallelogram at −52 °C.
Our motivation for the synthesis of crystalline CTNMOF was to explore the mechanical properties of metal–organic crystals with densely catenated backbones. Encouraged by the dynamic and adaptive nature of crystalline CTNMOF described above, we investigated its deformation properties by means of nanoindentation. To confirm the maximum load that can be applied to the crystal for the nanoindentation test, CTNMOF was subjected to a fracture test, where a fatal fracture of the crystal occurred when a load of 33 mN was applied to a CTNMOF single crystal (Supplementary Fig. 30). For the nanoindentation test, a single crystal of CTNMOF was placed on a low melting-point (47 °C) alloy firmly attached to a glass plate (Supplementary Fig. 29a). Because the CTNMOF crystal adopted a thin rectangular shape, only pressing along the a/b axes was technically possible. We pressed the crystal surface at 30 °C using a triangular pyramidal Berkovich diamond tip and released to obtain the loading and unloading mechanical features (Supplementary Fig. 32, Supplementary Table 17b), respectively (Fig. 4a, red trace). From the unloading curve, the Young’s modulus of CTNMOF was determined to be 6.31 ± 0.14 GPa (Fig. 4a, red trace). Although the observed Young’s modulus was much smaller than those of representative metal–organic crystals (Supplementary Table 19b), such as HKUST-143 (Fig. 4c, red trace, 10.65 ± 0.11 GPa (reported: 9.3 GPa)44), it is obviously larger than that of Zn-MOF (Zn2(NO2-BDC)2(DABCO) ⊃ DMF (ref. 45), where NO2-BDC is 2-nitro-1,4-benzene dicarboxylate and DABCO is 1,4-diazabicyclo[2.2.2]octane), the most elastic metal–organic crystal reported so far (Fig. 4e, red trace, 2.04 ± 0.26 GPa (reported: 2.1 GPa)45). This result is not disappointing, considering that the catenated macrocycles in CTNMOF are hydrogen-bonded with each other in an inter-component manner. Therefore, we soaked CTNMOF in DMF for 60 min to allow these hydrogen bonds to be attenuated. In fact, the vibrational band due to the hydrogen-bonded amide moieties of CTNMOF showed a 9 cm−1 shift from 1,631 cm−1 in air to 1,640 cm−1 in DMF (Supplementary Fig. 37). Then, DMF-including CTNMOF was subjected to the nanoindentation test in DMF (Supplementary Fig. 29b), where the unloading curve, in contrast to that in air (Fig. 4a, red trace), nearly returned to the origin (Fig. 4a, blue trace). As expected from this elastic nature, the Young’s modulus of CTNMOF in DMF was determined to be 1.77 ± 0.16 GPa (Supplementary Table 17a), which is very small and comparable only to that of polypropylene46 (1.5–2.0 GPa, Supplementary Table 19a). By contrast, the Young’s moduli of HKUST-1 (Supplementary Table 20) evaluated in air (Fig. 4c, red trace, Young’s modulus: 10.65 ± 0.11 GPa) and DMF (Fig. 4c, blue trace, Young’s modulus: 10.24 ± 0.33 GPa) differed only slightly from each other. Also, air-dried Zn-MOF (Fig. 4e, red trace, Young’s modulus: 2.04 ± 0.26 GPa), when soaked in DMF, even became 1.5 times less deformable (Fig. 4e, blue trace, Young’s modulus: 3.03 ± 0.35 GPa, Supplementary Table 21). When HKUST-1 (Fig. 4d) and Zn-MOF (Fig. 4f) were pressed, clear indentation footprints were left, as usual, on their surfaces. However, to our surprise, no indentation was left behind on the pressed parts of the CTNMOF surface (Fig. 4b). This observation may reflect the elastic nature of CTNMOF.

a, Load–displacement curves for CTNMOF in DMF (blue trace) and air-dried CTNMOF (red trace). The average Young’s moduli (E) in DMF (blue) and in air (red) are shown. The average Young’s moduli with standard deviation were evaluated using seven and eighteen measurement data points in DMF and in air, respectively. b, Micrograph of the surface of single-crystalline CTNMOF after the nanoindentation test in DMF. c, Load–displacement curves for HKUST-1 in DMF (blue trace) and air-dried HKUST-1 (red trace). The average Young’s moduli in DMF (blue) and in air (red) are shown. The average Young’s moduli with standard deviation were evaluated using eight and five measurement data points in DMF and in air, respectively. d, Micrograph of the surface of single-crystalline HKUST-1 after the nanoindentation test in DMF. e, Load–displacement curves for Zn-MOF in DMF (blue trace) and air-dried Zn-MOF (red trace). The average Young’s moduli in DMF (blue) and in air (red) are shown. The average Young’s moduli with standard deviation were evaluated using five and five measurement data points in DMFand in air, respectively. f, Micrograph of the surface of single-crystalline Zn-MOF after the nanoindentation test in DMF. The white dashed boxes in b, d, f indicate the locations tested for nanoindentation. g, Changes in the crystallographic parameters of CTNMOF upon applying high pressures (0–0.88 GPa). Scale bars, 50 µm.
Upon immersion in THF of DMF-including as-synthesized CTNMOF, we successfully obtained THF-including CTNMOF (Supplementary Fig. 28), whose load–displacement curve indicated an elastic nature with the Young’s modulus of 1.63 ± 0.13 GPa (Supplementary Fig. 34). Again, the vibrational band due to the hydrogen-bonded amide moieties of CTNMOF showed a 13 cm−1 shift from 1,631 cm−1 in air to 1,644 cm−1 in THF (Supplementary Fig. 37). Upon immersion in an additional 15 hydrogen bond competing and non-competing solvents, as-synthesized CTNMOF cracked, thereby making the nanoindentation test impossible. Therefore, we performed molecular dynamics simulations for a [2]catenane in hexane (Supplementary Figs. 44 and 45) as a representative of non-hydrogen bond competing solvents as well as in DMF (Supplementary Figs. 42 and 43) and gas phase (Supplementary Figs. 46 and 47), to explore how the hydrogen bonds between the catenated macrocycles in gas phase are modulated by solvation. The results clearly indicate that the hydrogen bonds are not attenuated in hexane but partially cleaved in DMF.
In relation to the above-mentioned nanoindentation experiments (Supplementary Fig. 31), we tested the behaviour of CTNMOF by applying a high isotropic pressure in a hydrostatic medium using a diamond anvil cell47,48. For this test, a single crystal of DMF-containing CTNMOF was placed in a diamond anvil cell with a diameter of 400 µm (Supplementary Fig. 26), and pressure-induced structural changes were investigated by in situ SCXRD. Unexpectedly, this elastic porous crystal was the most deformable along the c axis, rather than the a/b axes tested for nanoindentation: upon compression up to 0.88 GPa, 5% contraction occurred along the c axis without structural deterioration, whereas less than 0.4% contraction occurred along the a/b axes (Fig. 4g, Supplementary Tables 13–15). The crystal structure obtained at 0.46 GPa revealed that the observed anisotropic structural change was caused by the mutual sliding motions of the catenated macrocycles along the c axis, where the distance between the interlocked arms was extended by about 3%, resulting in 5% anisotropic contraction along the c axis (Supplementary Table 14 and Supplementary Fig. 27). Meanwhile, in THF, we found that THF-including CTNMOF showed a pressure-induced anisotropic deformation analogous to DMF-including CTNMOF in DMF (Supplementary Table 16) despite a smaller level of deformation than that of DMF-including CTNMOF in DMF. For spectroscopic understanding of this pressure-induced deformation, we carried out in situ Fourier transform infrared) spectroscopy of CTNMOF to investigate how the applied pressure affects the behaviour of the NHCO hydrogen bonds in the catenated macrocycles. For the guest-including CTNMOF, we chose the one including THF (Supplementary Fig. 28), rather than DMF, because included DMF interferes with the Fourier transform infrared spectroscopy. As shown in Supplementary Fig. 38a, the vibrational band due to the hydrogen-bonded NHCO groups showed an expected shift to a higher wavenumber region upon increasing the applied pressure to 2.4 GPa. By contrast, dried CTNMOF, upon being pressurized up to 2.1 GPa, did not show any shift in the NHCO vibrational band (Supplementary Fig. 38b).
Although the first polycatenane and polyrotaxane were synthesized more than 40 years ago, mechanical properties of such MIM-based materials have been little explored to date, because 3D materials that are densely composed of MIMs are very rare36. CTNMOF, as one of the first backbone-catenated 3D metal–organic crystals39, allows for switching between the mechanically rigid and elastic states by solvent-assisted modulation of the H-bonding lock of the MIM parts. If the elastic properties can be further enhanced by elaborating the MIM design, a squeezable MOF may be realized that can address the dilemma of porous materials where highly affinitive guests are not easily released.
Methods
SCXRD
Analysis for H4CTNL and Me4CTNL was performed using a Rigaku model VariMax Dual diffractometer equipped with a fine-focus sealed-tube X-ray source (λ = 0.71075 Å) and a confocal graphite monochromator. Data were collected and processed using CrystalClear (Rigaku)49. The analysis for CTNMOF was performed using a Rigaku XtaLAB P200 diffractometer with Mo-Kα radiation. Data were collected and processed using CrysAlisPro (Rigaku Oxford Diffraction)50. All data were corrected for Lorentzian and polarization effects. The structure was solved by direct methods51,52 expanded using Fourier techniques. The non-hydrogen atoms were refined anisotropically. Hydrogen atoms were refined using the riding model. The final cycle of full-matrix least-squares refinement (SHELXL97: ∑w(Fo2 − Fc2)2 where w is least-squares weights) on F2 was converged (the largest parameter shift was 0.00 times its estimated standard deviation (esd)) with unweighted and weighted agreement factors calculated based on the following equations: R1 = ∑||Fo| − |Fc||/∑|Fo| and wR2 = {∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]}½. Neutral atom scattering factors were taken from table 6.1.1.4 of ref. 53. Anomalous dispersion effects were included in Fcalc (calculated structure factor) (ref. 54); the values for ∆f′ (the real part of the anomalous-scattering factor) and ∆f″ (the imaginary part of the anomalous-scattering factor) were taken from table 4.2.6.8 of ref. 55. The values for the mass attenuation coefficients were taken from table 4.2.4.3 of ref. 56. All calculations were performed using the CrystalStructure57 crystallographic software package, except for refinement, which was performed using SHELXL9758. The heavily disordered solvent molecules were not modelled, and the SQUEEZE protocol inside PLATON41 was used to remove the void electron density at the end of the structure refinement. The crystallographic data have been deposited with the Cambridge Crystallographic Data Center (CCDC) (2080330–2080336, 2080245–2080247, 2080270). Details about structures and their corresponding CCDC numbers are listed in Supplementary Table 2.
PXRD
PXRD measurements were carried out using a Rigaku model RINT-ULTIMA III X-ray powder diffractometer equipped with a Cu sealed tube (λ = 1.54056 Å), which was operated at 40 kV and 40 mA. The PXRD data were collected in a 2θ range of 3° to 30° by using a step-scan mode with a step size of 0.02°. The powder sample was placed in a borosilicate glass capillary (WJM-Glas/Müller) with a diameter of 0.5 mm. For experiments shown in Fig. 2a–c, a small amount of the sample was taken for PXRD measurement at each step, namely, pristine, heated in vacuo and resoaked. For the experiments shown in Fig. 2d, e, all of the spectra were obtained from the same sample within a capillary. The solvent was added to/removed from the sample by centrifugation (3,000g, 2 min).
Thermogravimetric analysis
Thermogravimetric analysis was conducted using a Mettler-Toledo model TGA/SDTA851e in a temperature range from 25 °C to 600 °C at a heating rate of 1 °C min−1 under N2 with a flow rate of 20 ml min−1.
Gas sorption
A CO2 (99.995%) sorption experiment was conducted by using an automatic volumetric adsorption apparatus (BELSORP-max12-G-SP; MicrotracBEL) equipped with a cryostat system at −78 °C. A weighed sample (26 mg) of powdery CTNMOF was placed into a metallic copper cell and then kept under high vacuum for 6 h at 80 °C to remove the guest molecules. The sample cell was then placed into a cryostat system and evacuated at <10−2 Pa. The internal pressure change was monitored, and the amount of CO2 adsorbed was evaluated at each equilibrium point.
Nanoindentation
Experiments were carried out using an ENT-NEXUS (ELIONIX) instrument that is capable of performing nanoindentation tests in both dry and liquid environments. Nanoindentations were performed under the load-control mode at 30 °C. The CTNMOF crystals were fixed onto a glass plate using a Bi-based fusible alloy (Osaka Asahi U-47) to rigidly fix the specimens, even in DMF or THF. A diamond indenter tip with a Berkovich geometry was used. A maximum load value of 15 mN (except for Zn-MOF, where a maximum load of 8 mN (ref. 44) was applied) and a loading/unloading rate of 1.5 mN s−1 was used for all of the experiments. The holding time at the maximum load was set to be 5 s. The indentation moduli were obtained from the load–displacement curves using the standard Oliver–Pharr method59, where we assumed a Poisson’s ratio of 0.433 for the specimen44.
In situ high-pressure SCXRD
In situ SCXRD measurements under high pressure were performed using a Rigaku model Synergy custom diffractometer with a hybrid photon-counting detector (HyPix-6000HE). The X-ray source was a rotating Mo anode (λ = 0.7107 Å, Micromax-007), and the incident X-ray was focused on the sample position using a Rigaku model VariMax Mo confocal mirror. The single-crystal sample was loaded into a diamond anvil cell made of CuBe alloy equipping Boehler-Almax-type diamond anvils with a culet size of 800 μm and an opening angle of 40°. The gasket was a plate of CuBe alloy with a hole of 400 μm diameter and 200 μm thickness. DMF or THF was used as a pressure-transmitting medium. The sample pressures were estimated by a ruby fluorescence method60. The diffraction data were analysed using CrysAlisPro (Rigaku Oxford Diffraction)50 and the obtained cell parameters.
In situ high-pressure Fourier transform infrared spectroscopy
THF-containing CTNMOF and air-dried CTNMOF were used. THF-containing CTNMOF was prepared from as-synthesized CTNMOF by washing the crystals with THF several times and then soaking in THF for 24 h. CTNMOF and KBr were ground to fine powder in a mortar with a pestle. When THF-containing CTNMOF was used, THF was added to the sample mixture to prevent it from drying out. For THF-containing CTNMOF, an Inconel gasket was used with a hole of 300 μm diameter and 100 μm thickness. The gasket used for air-dried CTNMOF was a plate of CuBe alloy with a hole of 400 μm diameter and 200 μm thickness. A pair of Boehler-Almax-type conical diamonds with 400 μm culet were used as anvils. The sample pressures were estimated by a ruby fluorescence method60. Spectra were collected using a Bruker Hyperion 1000 infrared microscope.
Data availability
All data are available in the Article or its Supplementary Information.
References
Agam, G., Graiver, D. & Zilkha, A. Studies on the formation of topological isomers by statistical methods. J. Am. Chem. Soc. 98, 5206–5214 (1976).
Ogata, N., Sanui, K. & Wada, J. Novel synthesis of inclusion polyamides. J. Polym. Sci. Polym. Lett. Ed. 14, 459−462 (1976).
Raymo, F. M. & Stoddart, J. F. Interlocked macromolecules. Chem. Rev. 99, 1643−1663 (1999).
Bhushan, B. & Li, X. A review of nanoindentation continuous stiffness measurement technique and its applications. Mater. Charact. 48, 11–36 (2002).
Tan, J. C. & Cheetham, A. K. Mechanical properties of hybrid inorganic–organic framework materials: establishing fundamental structure–property relationships. Chem. Soc. Rev. 40, 1059–1080 (2011).
Bruns, C. J. & Stoddart, J. F. The Nature of the Mechanical Bond: From Molecules to Machines Ch. 1 (John Wiley & Sons, 2017).
Frisch, H. L. & Wasserman, E. Chemical topology. J. Am. Chem. Soc. 83, 3789–3795 (1961).
Evans, N. H. & Beer, P. D. Progress in the synthesis and exploitation of catenanes since the millennium. Chem. Soc. Rev. 43, 4658–4683 (2014).
Gil-Ramírez, G., Leigh, D. A. & Stephens, A. J. Catenanes: fifty years of molecular links. Angew. Chem. Int. Ed. 54, 6110–6150 (2015).
Xue, M., Yang, Y., Chi, X., Yan, X. & Huang, F. Development of pseudorotaxanes and rotaxanes: from synthesis to stimuli-responsive motions to applications. Chem. Rev. 115, 7398–7501 (2015).
Sauvage, J.-P. EurJOC—50 years of rotaxanes. Eur. J. Org. Chem. 2019, 3287–3288 (2019).
Amabilino, D. B. & Stoddart, J. F. Interlocked and intertwined structures and superstructures. Chem. Rev. 95, 2725–2828 (1995).
Sauvage, J.-P. & Dietrich-Buchecker, C. Molecular Catenanes, Rotaxanes and Knots: a Journey Through the World of Molecular Topology (Wiley-VCH, 1999).
Kassem, S. et al. Artificial molecular motors. Chem. Soc. Rev. 46, 2592–2621 (2017).
Erbas-Cakmak, S., Leigh, D. A., McTernan, C. T. & Nussbaumer, A. L. Artificial molecular machines. Chem. Rev. 115, 10081–10206 (2015).
Niu, Z. & Gibson, H. W. Polycatenanes. Chem. Rev. 109, 6024–6046 (2009).
Takata, T. Polyrotaxane and polyrotaxane network: supramolecular architectures based on the concept of dynamic covalent bond chemistry. Polym. J. 38, 1–20 (2006).
Harada, A., Hashidzume, A., Yamaguchi, H. & Takashima, Y. Polymeric rotaxanes. Chem. Rev. 109, 5974–6023 (2009).
Wu, Q. et al. Poly[n]catenanes: synthesis of molecular interlocked chains. Science 358, 1434–1439 (2017).
Mena-Hernando, S. & Pérez, E. M. Mechanically interlocked materials. Rotaxanes and catenanes beyond the small molecule. Chem. Soc. Rev. 48, 5016–5032 (2019).
Qiu, Y. et al. A precise polyrotaxane synthesizer. Science 368, 1247–1253 (2020).
Okumura, Y. & Ito, K. The polyrotaxane gel: a topological gel by figure‐of‐eight cross‐links. Adv. Mater. 13, 485–487 (2001).
Choi, S., Kwon, T.-W., Coskun, A. & Choi, J. W. Highly elastic binders integrating polyrotaxanes for silicon microparticle anodes in lithium ion batteries. Science, 357, 279–283 (2017).
http://www-materials.eng.cam.ac.uk/mpsite/properties/non-IE/stiffness.html
Charitidis, C. A., Koumoulos, E. P., Nikolakis, V. & Dragatogiannis, D. A. Structural and nanomechanical properties of a zeolite membrane measured using nanoindentation. Thin Solid Films 526, 168–175 (2012).
Kitagawa, S., Kitaura, R. & Noro, S. Functional porous coordination polymers. Angew. Chem. Int. Ed. 43, 2334–2375 (2004).
Yaghi, O. M. et al. Reticular synthesis and the design of new materials. Nature 423, 705–714 (2003).
Loeb, S. J. Metal–organic rotaxane frameworks; MORFs. Chem. Commun. 1511–1518 (2005).
Li, Q. et al. A catenated strut in a catenated metal–organic framework. Angew. Chem. Int. Ed. 49, 6751–6755 (2010).
Li, Q. et al. A metal–organic framework replete with ordered donor–acceptor catenanes. Chem. Commun. 46, 380–382 (2010).
Zhao, Y.-L. et al. Rigid-strut-containing crown ethers and [2]catenanes for incorporation into metal–organic frameworks. Chem. Eur. J. 15, 13356–13380 (2009).
Vukotic, V. N., Harris, K. J., Zhu, K., Schurko, R. W. & Loeb, S. J. Metal–organic frameworks with dynamic interlocked components. Nat. Chem. 4, 456–460 (2012).
Zhu, K., Vukotic, V. N., O’Keefe, C. A., Schurko, R. W. & Loeb, S. J. Metal–organic frameworks with mechanically interlocked pillars: controlling ring dynamics in the solid-state via a reversible phase change. J. Am. Chem. Soc. 136, 7403–7409 (2014).
Zhu, K., O’Keefe, C. A., Vukotic, V. N., Schurko, R. W. & Loeb, S. J. A molecular shuttle that operates inside a metal–organic framework. Nat. Chem. 7, 514–519 (2015).
Chen, Q. et al. A redox-active bistable molecular switch mounted inside a metal–organic framework. J. Am. Chem. Soc. 138, 14242–14245 (2016).
Frank, N. C., Mercer, D. J. & Loeb, S. J. An interwoven metal-organic framework combining mechanically interlocked linkers and interpenetrated networks. Chem. Eur. J. 19, 14076–14080 (2013).
Gholami, G., Zhu, K., Ward, J. S., Kruger, P. E. & Loeb, S. J. Formation of a polythreaded, metal–organic framework utilizing an interlocked hexadentate, carboxylate linker. Eur. J. Inorg. Chem. 4524–4529 (2016).
Gholami, G., Baggi, G., Zhu, K. & Loeb, S. J. Metal–organic frameworks utilising an interlocked, hexadentate linker containing a tetra-carboxylate axle and a bis(pyridine) wheel. Dalton Trans. 46, 2462–2470 (2017).
Lewis, J. E. M. Self-templated synthesis of amide catenanes and formation of a catenane coordination polymer. Org. Biomol. Chem. 17, 2442–2447 (2019).
Johnston, A. G., Leigh, D. A., Pritchard, R. J. & Deegan, M. D. Facile synthesis and solid-state structure of a benzylic amide [2]catenane. Angew. Chem. Int. Ed. Engl. 34, 1209–1212 (1995).
Spek, A. L. Single-crystal structure validation with the program PLATON. J. Appl. Cryst. 36, 7–13 (2003).
Schneemann, A. et al. Flexible metal–organic frameworks. Chem. Soc. Rev. 43, 6062–6096 (2014).
Chui, S. S.-Y., Lo, S. M.-F., Charmant, J. P. H., Orpen, A. G. & Williams, I. D. A chemically functionalizable nanoporous material [Cu3(TMA)2(H2O)3]n. Science 283, 1148–1150 (1999).
Bundschuh, S. et al. Mechanical properties of metal–organic frameworks: an indentation study on epitaxial thin films. Appl. Phys. Lett. 101, 101910 (2012).
Henke, S., Li, W. & Cheetham, A. K. Guest-dependent mechanical anisotropy in pillared-layered soft porous crystals—a nanoindentation study. Chem. Sci. 5, 2392–2397 (2014).
Engineering ToolBox. Young’s modulus—tensile and yield strength for common materials, https://www.engineeringtoolbox.com/young-modulus-d_417.html (2003).
Boehler, R. New diamond cell for single-crystal X-ray diffraction. Rev. Sci. Instrum. 77, 115103 (2006).
Ratajczyk, P., Sobczak, S. & Katrusial, A. High-pressure structure and properties of N,N-dimethylformamide. Cryst. Growth Des. 19, 896–901 (2019).
CrystalClear v.1.3.6 (Rigaku, 2015).
CrysAlis v.40.84a (Rigaku, 2015).
Burla, M. C. et al. SIR2011: a new package for crystal structure determination and refinement. J. Appl. Cryst. 45, 357–361 (2012).
Burla, M. C. et al. IL MILIONE: a suite of computer programs for crystal structure solution of proteins. J. Appl. Cryst. 40, 609–613 (2007).
Creagh, D. C. & McAuley, W. J. in International Tables for Crystallography Vol. C (ed. Wilson, A. J. C.) 572 (Kluwer Academic, 1992).
Ibers, J. A. & Hamilton, W. C. Dispersion corrections and crystal structure refinements. Acta Crystallogr. 17, 781–782 (1964).
Creagh, D. C. & McAuley, W. J. in International Tables for Crystallography Vol. C (ed. Wilson, A. J. C.) 219–222 (Kluwer Academic, 1992).
Creagh, D. C. & Hubbell, J. H. in International Tables for Crystallography, Vol. C (ed. Wilson, A. J. C.) 200–206 (Kluwer Academic, 1992).
CrystalStructure v.4.2.5 (Rigaku, 2017).
Sheldrick, G. A. A short history of SHELX. Acta Crystallogr. A 64, 112–122 (2008).
Oliver, W. C. & Pharr, G. M. Measurement of hardness and elastic modulus by instrumented indentation: advances in understanding and refinements to methodology. J. Mater. Res. 19, 3–20 (2004).
Piermarini, G. J., Block, S. J., Barnett, D. & Forman, R. A. Calibration of the pressure dependence of the R1 ruby fluorescence line to 195 kbar. J. Appl. Phys. 46, 2774–2780 (1975).
Acknowledgements
A part of this work was conducted at the Advanced Characterization Nanotechnology Platform of the University of Tokyo, supported by ‘Nanotechnology Platform’ of the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan. We thank K. Okitsu and K. Fukawa for technical assistance with SCXRD and PXRD, respectively; D. Hamane for discussions on high-pressure experiments; T. Uemura, N. Hosono and H. Taketomi for help with thermogravimetry/mass spectrometry measurements; S. Suginome for help with NMR spectral measurements and J.-S. M. Lee and K. Morishita for discussion on preparing the manuscript. T.A. acknowledges the Japan Society for the Promotion of Science (JSPS) for a JSPS Grant-In-Aid for Scientific Research (S) (18H05260) on ‘Innovative Functional Materials based on Multi-Scale Interfacial Molecular Science’. This work was also supported by JST, PRESTO (grant number JPMJPR20A5), Japan. H.S. is grateful for JSPS KAKENHI grant numbers 17H05357 (Coordination Asymmetry), 18H04501 (Soft Crystal) and 20H02705 (Scientific Research (B)). W.M. acknowledges the receipt of a JSPS Postdoctoral Fellowship for Research in Japan and JSPS KAKENHI grant number 20K15251.
Author information
Authors and Affiliations
Contributions
W.M., H.S. and T.A. conceived the project, designed experiments and directed the research. W.M. performed and interpreted all of the experiments associated with molecular synthesis, crystal growth and structural characterization. H.S. performed the sorption experiments. S.K., W.M. and Y.I. designed nanoindentation experiments. S.K., T.I. and W.M. conducted nanoindentation experiments. K.K., W.M. and H.S. performed high-pressure experiments. J.P. and Y.H. conducted the computational studies. All authors contributed to the writing and editing of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
This file contains Supplementary text, figs. 1–47, tables 1–23 and references.
Rights and permissions
About this article

Cite this article
Meng, W., Kondo, S., Ito, T. et al. An elastic metal–organic crystal with a densely catenated backbone. Nature 598, 298–303 (2021). https://doi.org/10.1038/s41586-021-03880-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-021-03880-x
This article is cited by
-
Unconventional mechanical and thermal behaviours of MOF CALF-20
Nature Communications (2024)
-
Light-driven anisotropy of 2D metal-organic framework single crystal for repeatable optical modulation
Communications Materials (2024)
-
Elastic hydrogen-bonded ionic framework
Nano Research (2023)
-
Controlling dynamics in extended molecular frameworks
Nature Reviews Chemistry (2022)
-
Controllable photomechanical bending of metal-organic rotaxane crystals facilitated by regioselective confined-space photodimerization
Nature Communications (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
