
Abstract
Deletions on chromosome 15q14 are a known chromosomal cause of cleft palate, typically co-occurring with intellectual disability, facial dysmorphism, and congenital heart defects. The identification of patients with loss-of-function variants in MEIS2, a gene within this deletion, suggests that these features are attributed to haploinsufficiency of MEIS2. To further delineate the phenotypic spectrum of the MEIS2-related syndrome, we collected 23 previously unreported patients with either a de novo sequence variant in MEIS2 (9 patients), or a 15q14 microdeletion affecting MEIS2 (14 patients). All but one de novo MEIS2 variant were identified by whole-exome sequencing. One variant was found by targeted sequencing of MEIS2 in a girl with a clinical suspicion of this syndrome. In addition to the triad of palatal defects, heart defects, and developmental delay, heterozygous loss of MEIS2 results in recurrent facial features, including thin and arched eyebrows, short alae nasi, and thin vermillion. Genotype–phenotype comparison between patients with 15q14 deletions and patients with sequence variants or intragenic deletions within MEIS2, showed a higher prevalence of moderate-to-severe intellectual disability in the former group, advocating for an independent locus for psychomotor development neighboring MEIS2.
Similar content being viewed by others


Introduction
Orofacial clefts are the most common craniofacial human birth defect, affecting the lip, both the lip, and the palate or the palate alone (CP). The spectrum of palatal clefts ranges from subclinical phenotypes like bifid uvula and high-arched palate, to submucous CP and velopharyngeal insufficiency, and to overt cleft palate. Most palatal clefts are isolated and sporadic, and are considered to have a multifactorial cause. Orofacial clefts, associated with developmental delay, dysmorphic features, or other major congenital anomalies, are defined syndromic. These mostly have a single genetic cause, either chromosomal or monogenic.
Deletions on chromosome 15q14 are a known chromosomal cause of cleft palate, typically co-occurring with intellectual disability (ID), facial dysmorphism and congenital heart defects (CHD) [1,2,3,4,5,6,7,8,9]. De novo loss-of-function variants in MEIS2, a gene within this region, were previously described in two patients with CP, ID, and CHD [10, 11]. Therefore, these features are attributed to haploinsufficiency of MEIS2. The MEIS genes belong to the three-amino-acid-loop extension (TALE) superfamily of homeobox (HOX) genes, highly preserved transcription factors consisting of a HOX with a helix-turn-helix structure of 60 amino acids, extended by a TALE between alpha helices 1 and 2 in the homeodomain. MEIS2 most likely functions as a HOX co-factor, which binds to Pbx proteins and/or HOX proteins to form dimeric or trimeric complexes to enhance the specificity and affinity of DNA binding. MEIS2 is expressed during early fetal brain development in humans [12]. In zebrafish, Meis2 has been proven important in development of the mesencephalon [13], the craniofacial skeleton [14], and the heart [15, 16]. Conditional knockout of Meis2 in developing murine neural crest cells results in abnormalities of the craniofacial skeleton, cranial nerves, and heart [17]. Results from animal models are in line with the triad of CHD, palatal defects and ID, previously observed in patients with variants or deletions of MEIS2.
To further delineate the phenotypic spectrum of this MEIS2-related syndrome, we collected 23 previously unreported patients either with a de novo variant in MEIS2 (patients 1–9), or with a 15q14 microdeletion encompassing this gene (patients A to N). Genotype–phenotype comparison was undertaken to identify recurrent features of the 15q14 deletion syndrome, which are not related to MEIS2, but rather to one of the neighboring genes.
Case reports
Detailed clinical reports of patients 1 to 9 are provided below. An overview of the features of the other patients (A to N) can be found in Table 2.
Patient 1
This boy is the second child of healthy, unrelated parents of North African descent. Familial history was negative with regard to developmental delay or congenital malformations. He was born at 39 weeks of gestation with weight 3.150 kg (25–50th centile), length 48 cm (10–25th centile) and head circumference 32 cm (below 3rd centile). At birth, cleft palate and a small perimembranous ventricular septal defect (VSD) were diagnosed. Hearing was normal. Clinical examination revealed proptosis and a downslant of the palpebral fissures. There was a mild eversion of the lower eyelids, fine arched eyebrows, and a small chin (Fig. 1). The metopic suture was prominent. At the age of 5 year 3 months, weight and height were at the 25th centile and head circumference was below the 3rd centile (−2.1 SD). The VSD had not closed spontaneously at that age. Psychomotor development was delayed. He walked at age 22 months. At age 3 years 5 months, non-verbal IQ was 64 (SON-R). Both fine and gross motor development were equivalent to the level of a child aged 2 years and 10 months. Speech was even more delayed, lagging behind more than 1.5 years.

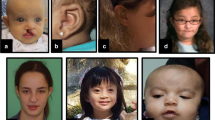
Facial phenotypes from patients with de novo variants in MEIS2 (patients 1–6, 8, and 9) and from patient K and L, who were identified with a de novo deletion on 15q14. Numbers or letters in the left upper corner refer to the patient numbering in Tables 1 and 2. Although facial phenotypes are variable and no clinically recognizable facial gestalt can be delineated, some recurrent features were noticeable, including thin, arched eyebrows, a metopic ridge, a thin vermillion, and short alae nasi. The bottom row shows evolution of facial features with age in patient K (from the age of 8 months to the age of 28 years). Note the thin, arched, and laterally displaced eyebrows, thin upper lip and broad nasal root and tip with short alae nasi, as well as facial coarsening with age
Patient 2
This boy was born from healthy, unrelated parents of Caucasian origin. At birth he presented with cleft palate and gastro-esophageal reflux. He developed conductive hearing loss and required bilateral hearing aids. He had sleep apnea, treated by nocturnal continuous positive airway pressure. This boy was referred to the genetics clinic because of developmental delay and behavioral problems. He started walking at the age of 18 months. His speech was moderately delayed with a predominant speech sound disorder with hypernasality. He was diagnosed with autism spectrum disorder and presented with temper tantrums, aggressive behavior, and short attention span. Brain MRI was normal, except for mild asymmetry of the lateral ventricles. At age 10 years 11 months, his height was 145.3 cm (50th centile), weight 40.5 kg (75th centile) and head circumference 56.5 cm (75–90th centile). Facial dysmorphic features included ptosis of the left eye, sagging of the lower eyelids, and square-shaped ear helices (Fig. 1).
Patient 3
Patient 3 is a 4-year-old boy, who was born full term as the first child of healthy, unrelated parents of Caucasian descent. The pregnancy was uncomplicated. He was described as an irritable baby and required syringed feeds for the first 3 days of life owing to poor suck. He has been managed for submucous cleft palate, hypermetropia, strabismus, and cryptorchidism. He was referred to the genetics clinic with autism spectrum disorder and developmental delay. He started walking independently from 30 months of age and had difficulties with co-ordination. He was not able to run or jump. He had 4–5 word sentences and understood simple instructions. His head circumference was 49.6 cm (5th centile). His weight and length were at the 25th centile. He presented with facial dysmorphism, featuring epicanthic folds, hypoplastic alae nasi, prominent ears, and a frontal cow lick (Fig. 1).
Patient 4
Patient 4 is a 20-year-old young woman of Caucasian descent, the second of four children. Pregnancy was uncomplicated. Delivery was at 42 weeks of gestation by cesarean section. Her birth weight was 4.5 kg. A median cleft palate was observed. After introduction of a special needs feeder, feeding was adequate. The cleft palate was surgically corrected at age 10 months. At the age of 6 months, she presented with limited movement with intermittent hyperextension of the trunk. Her muscular tone was normal. At age 12 months she was diagnosed with mild myopia with adequate vision. Hearing was intermittently compromised by middle ear infections. At age 14 months her motor and cognitive development was 6 months delayed. She could sit and walk unassisted, respectively, from the age of 2 and 3 years. She made sounds and spoke a few words at age 18 months; speech development was severely delayed. She attended a specialized school for children with compromised speech development. At age 20 years, however, speech is adequate, and in line with her cognitive development, which is mildly impaired.
Current biometric parameters are within normal range, with height being 173 cm (50th centile), weight 50 kg (10–25th centile), and head circumference 54.5 cm (25th centile). She has a broad forehead with bitemporal narrowing. Her limbs were normal, apart from diminished pronation and supination of the forearms. Brain MRI (at age 3 years), metabolic investigations, X-rays of the arms and hands (at age 2 years) were normal. Cardiologic investigations at age 20 years showed minimal billowing and insufficiency of the mitral valve.
Patient 5
This patient is a 14-year-old male of Mexican origin [18]. He was born at term following an uncomplicated pregnancy. Birth weight was 3.6 kg. He was noted to have cryptorchidism. Left testicle orchidopexy and inguinal hernia repair were performed at 1 year. Additional genitourinary surgeries included right-sided orchidopexy to correct retractile right testicle at 5 years and phimosis/meatal stenosis repair at 6 years. Bifid uvula and possible submucosal cleft palate were identified at 16 months. Developmentally, he started walking with support at 16 months and independently at 18 months. He started speaking at 2 years and was able to speak in 2–3 word sentences by the age of three and a half. Brain and lumbar spine MRI and EEG completed at that time were unremarkable. At 2 years of age he was noted to have several dysmorphic features including prominent forehead, epicanthic folds, hypertelorism, long eyelashes with distichiasis, bulbous beaked nose with short alae nasi, bifid uvula, thin upper lip, retrognathia, short neck, hypoplastic right nipple, and fifth finger clinodactyly. Cardiac evaluation at that age was unremarkable and a murmur was deemed to be physiologic. He was noted to have precocious adrenarche and evaluated by an endocrinologist at the age of 5. HCG stimulation test showed borderline/low testosterone levels after stimulation. Myringotomy tubes were placed at 6 years owing to a history of recurrent ear infections and possible hearing loss. Ocular melanocytosis and iris nevus were noted on ophthalmology evaluation at 8 years. He was also noted to have “pre-glaucoma” at the age of 10 years old, for which he is being monitored. Most recent clinical evaluation at 13 years was significant for broad forehead, atypical medial eyebrow flare, distichiasis, micrognathia, mild posterior rotation of ears, and bifid uvula (Fig. 1). His head circumference was 57 cm (97th percentile), his height was 173 cm (98th percentile), and his weight was 72.3 kg (97th percentile).
Patient 6
Patient 6 is the first child of healthy, unrelated parents of Caucasian origin. She was born at term after an uncomplicated pregnancy. At birth she presented with a cleft palate. No associated anomalies were noted. She has fine arched eyebrows and hypoplastic alae nasi, but she was not considered dysmorphic (Fig. 1). She presented with mild developmental delay. She started walking at 25 months of age. Speech delay was noted which required speech therapy. Aged 5 years 8 months, her height was 101 cm (3rd centile), weight 13.5 kg (below 3rd centile (−2.5 SD)) and head circumference 49.5 cm (10th centile). Physical examination was normal. She exhibits learning difficulties.
Patient 7
This 18-year-old female is of Asian-Caucasian origin. She was born with a complex congenital heart defect: tetralogy of Fallot with an Ebstein’s anomaly of the tricuspid valve. Early in life she had feeding difficulties, but neither palatal cleft, nor velopharyngeal insufficiency were reported. Psychomotor development was mildly delayed, requiring occupational therapies. At the age of 18 years, she was in the 11th grade of secondary school, without an individualized education program. She measured 160 cm (25th centile) and weighs 43.2 kg (3rd centile). She was not reported to be dysmorphic.
Patient 8
This girl was the first child of healthy, unrelated parents of Caucasian descent. The father had macrocephaly and the mother had a unilateral ear pit. Family history was otherwise unremarkable. The patient was born at 39 weeks of gestation. Birth weight was 2520 g (3rd centile), length was 48 cm (10–25th centile), and head circumference was 36 cm (90th centile). At birth, she presented with severe cyanosis, caused by an Ebstein’s anomaly of the tricuspid valve with a hypoplastic right ventricle, a 7-mm perimembranous ventricular septum defect and a secundum atrial septum defect. In addition, she presented with a cleft of the palatum molle and a subtle notch in the hard palate. The congenital heart defect was treated surgically at the age of 8 months and, subsequently at the age of 3 years, to install a total cavopulmonary connection. The soft palate was surgically closed and ear tubes were placed at the age of 14 months. The first months of life were complicated by severe feeding problems, requiring tube feeding via a gastrostomy. This girl presented at the genetics clinic at 17 months, and was noted to have dysmorphic facial features including a high and broad forehead, fine arched eyebrows, hypoplastic alae nasi, short philtrum, and low-set dysplastic ears with a right-sided ear pit (Fig. 1). Hands and feet were normal, except for bilaterally overriding toes. She was delayed in attaining early developmental milestones. She walked independently from the age of 23 months. Until the age of 10 years, she needed speech therapy because of articulation difficulties and hypernasal speech. Currently, she attends a special needs school for children with learning problems. Her height and weight are within normal range, both mapping at the 25th centile. Her head circumference is at the 97th centile.
Patient 9
Patient 9 was the first child of a first and dizygotic twin pregnancy of healthy, unrelated parents of Caucasian descent. Family history was unremarkable. Prenatally an atrial and VSD were detected; a double bubble gave the suspicion of a duodenal stenosis. He was born prematurely by cesarean section at 32 weeks due to pathological Doppler velocimetry indicating a centralization of fetal circulation. Birth weight was 1340 g (10–25th centile), birth length 38 cm (5th centile), and occipitofrontal circumference at birth 29 cm (25th centile). Immediately after birth he required respiratory assistance. On the second day of life, a duodenal membrane causing stenosis was corrected. The atrial and ventricular septal defects were confirmed postnatally. In addition, stenosis of the left lower pulmonary vein was detected. The heart defect resulted in a heart failure, requiring closure of the septal defects at the age of 5 months. Bilateral inguinal hernia required treatment at the age of 7 months. The leading problem of this boy was a permanent respiratory insufficiency that could not be explained solely by his heart defect and was likely caused by malfunctioning pulmonary ventilation. The boy could not be weaned from respiratory assistance. Over the course of the disease he required artificial ventilation and a tracheostomy was implemented at the age of 11 months. In addition, he developed hypothyroidism, transitory pancreatitis, and nephrocalcinosis. He suffered from feeding difficulties, recurrent vomiting and failure to thrive. Repeatedly he had high temperatures without infections, probably owing to a disturbed central temperature regulation. His neurological development was poor. Brain ultrasound was normal at birth. Repeated brain imaging later in life showed bilateral ventriculomegaly and brain atrophy. At the age of 13 months his weight was 7.73 kg (< 3rd centile), length 65 cm (< 3rd centile (− 3.5 SD) and head circumference 44 cm (< 3rd centile (− 2.2 SD). He presented with profound developmental delay, severe muscular hypotonia of the trunk and hypertonia of the limbs. He had poor head control and he did not roll over. He did not interact and did not grasp. He showed a strained breathing and a restless behavior. Dysmorphic facial features included high and small forehead with a high frontal hairline, frontal bossing and temporal narrowing, short palpebral fissures, hypertelorism, a depressed nasal bridge with anteverted nares, full cheeks, and a small mouth (Fig. 1). Neck and limbs were short in comparison with the trunk. The boy died from respiratory insufficiency at the age 13 months.
Methods
Genomic DNA from the patients and their parents was extracted from peripheral leukocytes of ethylenediaminetetraacetic acid-treated blood according to standard methods. All procedures involving patients performed in this study were in accordance with the ethical standards of their respective institutional research committees. Written consent was obtained from all patients or from their legal representatives. Analysis is based on GRCh37/hg19 assembly. Exons are numbered according to NG_029108.1. Identified MEIS2 variants were uploaded in the LOVD database (http://www.lovd.nl/MEIS2). Deletions, harboring MEIS2 are available in the Decipher database (https://decipher.sanger.ac.uk/).
Copy Number analysis
For patients 1–8 chromosomal microarray analysis was performed, according to the manufacturer’s instructions (supplementary methods). For patient 9, copy number variation was deduced from the whole-exome sequencing data (supplementary methods).
Next-generation sequencing (NGS)
Patients 1–7 and 9 were submitted to whole-exome sequencing (WES) for unexplained syndromic ID, CHD, and/or palatal defects. WES for patient 6 was restricted to the affected patient, followed by Sanger sequencing of potentially disease-causing variants in parental DNA. For all other patients NGS of parent-offspring trios was performed. For patient 2 and 3, WES was performed through the Deciphering Developmental Disorders (DDD) study, as described previously [19]. Patient 5 was enrolled in a research study at the Institute for Genomic Medicine at Columbia University (protocol AAAO8410). Patient 7 was recruited from the Pediatric Cardiac Genomics Consortium (PCGC) from the National Heart, Lung, and Blood Institute (NHLBI) and the Eunice Kennedy Shriver National Institute of Child Health and Human Development, and was analyzed as described by Homsy et al. [20]. A detailed overview of the applied NGS methods and variant annotation pipelines is provided as supplementary methods.
Sanger sequencing
For patient 8, targeted sequencing of all exons and exon–intron boundaries of the MEIS2 gene was performed based on phenotypical resemblance with previously described patients with MEIS2 variants. Sanger sequencing was performed for the 12 coding exons and exon–intron boundaries of the MEIS2 gene (NM_170674.4). Primers are available in supplementary table 1. The amplification products were purified, directly sequenced using universal primers with the BigDye terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Thermo Fischer Scientific, MA, USA), and analyzed on a 3730xl genetic analyzer (Applied Biosystems).
Sanger sequencing was applied in patients 1, 6, and 8, and their respective parents to confirm the de novo occurrence of the MEIS2 variant in a heterozygous state.
Genotype–phenotype correlation with 15q14 deletion patients
PubMed abstracts were searched for reported patients with 15q14 deletions, which were identified or delineated by chromosome microarray analysis. The following deletions were excluded from further analysis: (1) known copy number polymorphisms (CNV reported in at least 1% of normal population databases), (2) deletions larger than 7 Mb in size, (3) deletions extending beyond chromosomal band 15q14, or (4) deletions not containing (at least one exon of) the gene MEIS2.
Subsequently, our locally curated CNV database and the publicly available database DECIPHER [21] were searched for unpublished patients with 15q14 deletions, in line with the inclusion criteria enlisted above. Phenotype requests were sent to the responsible clinician. Deletions for which no accurate phenotyping could be retrieved, were excluded from further analysis. Genotype–phenotype comparison was performed between patients with 15q14 deletions and patients with de novo variants in MEIS2, to find recurrent features in the deletion cohort, which could not be ascribed to haploinsufficiency of the gene MEIS2, but rather to one of the neighboring genes.
Results
Copy number analysis in patients 1–9
No known disease-causing copy number changes were identified in patients 1–8, undergoing chromosomal microarray analysis. In patient 5, SNP array analysis rendered a rare maternally inherited 6q25.2 duplication (chr6:g.(?_153413459)_(154286581_?)dup), which was considered probably benign. Exome sequencing in patient 9 identified a de novo heterozygous deletion of 185 base pairs on chromosome 3p13 (chr3:g.(71179649_71179833)del), encompassing exon 1 of the less common FOXP1 transcription variant NM_001244815.1.
Identification of de novo MEIS2 variants
A de novo variant in the gene MEIS2 was identified in patients 1–9, either by WES, or by Sanger sequencing (three frameshift, two nonsense, three splice site, and one missense variant). Table 1 and Fig. 2 depict the positions of de novo variants in MEIS2 in this cohort, complemented by two previously reported MEIS2 variants [10, 11]. All de novo variants were absent in the 1000Genome (http://www.internationalgenome.org/), Exome Aggregation Consortium population databases (http://exac.broadinstitute.org/) and GnomAD databases (http://gnomad.broadinstitute.org/). The variants in patients 1–8 were predicted to cause protein truncation or nonsense-mediated decay, and therefore to cause loss-of-function. The missense variant in patient 9 was predicted to be damaging by in silico prediction software. The recently developed prediction tool REVEL (rare exome variant ensemble learner) yielded a score of 0.838, corresponding to a high specificity for affecting function [22]. In addition, this patient was found to be compound heterozygous for two rare variants of unknown significance in the gene LRP2, c.8624G>A, p.(Arg2875His), and c.10424G>C, p.(Gly3475Ala) (c.[8624G>A];[10424G>C]; NM_004525.2/NP_004516.2). His unaffected twin brother was heterozygous for one of these variants. Patients 1–8 were not found to carry additional de novo, X-linked, compound heterozygous or homozygous variants, which could explain or contribute to their phenotype.

Lollipop plot displaying novel and previously described de novo variants in MEIS2. Variants are plotted on a linear protein, using MutationMapper (cBioPortal–http://www.cbioportal.org/index.do) as described [34, 35]. Lollipops represent de novo protein changes in MEIS2. The dark and light gray box, respectively, represent the N-terminal of homeobox Meis/PKNOX1 and the homeobox KN domain (or TALE-containing homeodomain). The p.(Arg333Lys) missense variant in patient 9 affects the same residue, that was found deleted in the patient reported by Louw et al. [10]
Clinical data from patients 1–9 and both previously published patients are summarized in Table 1. Key features of the MEIS2-related syndrome include palatal defects (9/10; 90%), ranging from bifid uvula to overt CP, ID (100%), CHD (7/11; 63%). Development is typically mildly delayed, with moderate-to-severe ID occurring in 4 out of 11 patients (36%). Three patients were reported with autism spectrum disorder. No recognizable facial gestalt could be ascribed to this syndrome. However, recurrent facial features include thin, arched eyebrows, bitemporal narrowing, hypoplastic alae nasi, and a thin upper lip (Fig. 1).
Genotype–phenotype comparison with 15q14 deletion patients
Clinical and molecular details from 16 previously published families [1,2,3,4,5,6,7,8,9] and 15 novel patients (patients A to N, and Decipher patient 286841) with 15q14 deletions were collected (Fig. 3). Deletion sizes ranged from 123 kb to 6.97 Mb. Two deletions involved an intragenic deletion of MEIS2 [4, 9], whereas seven deletions only affected MEIS2 and its proximal neighbor C15orf41 (patient A and C, Decipher 286841, Johansson et al. [6], Gambin et al. [9]). Decipher patient 286841, as well as the six patients reported recently by Gambin et al. [9], were excluded from further genotype–phenotype analysis, as no sufficient clinical features could be retrieved (supplementary table 3). Twenty-five deletion carriers from 24 independent families were retained (patients A to N and 11 previously reported families [1,2,3,4,5,6,7,8]) (Table 2 and supplementary table 2). All 24 deletions affected the gene MEIS2, either completely (14/24), or partially (10/24). The deletion occurred de novo in 18 families and was inherited from an affected mother in one family (family 2 by Johansson et al. [6]). Inheritance was unknown in the five remaining families. Breakpoints of the deleted regions were unique, advocating for non-homologous end-joining as the underlying pathogenic mechanism of these 15q14 deletions. We also included one previously reported family with an intragenic tandem duplication in MEIS2 with four affected family members (family 1 by Johansson et al. [6]). This duplication was predicted to cause loss-of function.

Overview of 14 novel (patients A to N) and 17 previously reported deletions on chromosome 15q14, harboring at least one exon of the gene MEIS2. Deletions are depicted as red boxes. The intragenic duplication, reported in family 1 by Johansson et al., is depicted as a blue box [6]. The region corresponding to the genomic position of the MEIS2 gene is represented by a blue dotted box
In three families copy number analysis rendered an additional rare variant elsewhere in the genome. Patient A had a 500 kb duplication on 5p15.31 (chr5:g.(?_8747001)_(9247598_?)dup), and a 206 kb duplication on 5q15.2, harboring the gene CTNND2 (chr5:g.(?_11298758)_(11505375_?)dup). Both duplications were inherited from an unaffected mother. Intragenic deletions in CTNND2 are considered a cause for ID [23]. It is currently unclear whether this partial CTNND2 duplication occurred in tandem, and whether it contributed to the patient’s phenotype. In patient C a de novo deletion on chromosome 12q23.3 was identified (chr12:g.(?_108249783)_(108711392_?)del). This deletion is absent from patient and normal population databases and affects two genes (WSCD2 and CMKLR1), which are not yet linked to developmental disorders. The duplication on Xq25, detected in patient M, harbors the genes THOC2 and XIAP, but not STAG2. Loss-of-function variants in THOC2 and XIAP are respectively causing X-linked recessive ID [24] and lymphoproliferative syndrome [25]. Duplications on Xq25, encompassing XIAP and STAG2, were considered disease-causing in two families with X-linked ID and facial dysmorphism, including malar hypoplasia and prognathism [26]. Although not comprising STAG2, this duplication might have contributed to the severe ID and malar hypoplasia in patient M.
Discussion
We contribute to the delineation of the MEIS2-related human phenotype by adding 23 novel patients either with de novo variants (patients 1–9), or with deletions of this gene (patients A to N). Novel de novo variants in MEIS2 included two nonsense variants (patient 1 and 4), three frameshift variants (patients 5, 6, and 7), three predicted splice variants (patients 2, 3, and 6) and one missense variant (patient 9). ExAc constraint scores of MEIS2 for loss-of-function and missense variants are respectively 0.99 and 3.20, indicating intolerance of MEIS2 towards loss-of-function and missense variants. The missense variant was found in a severely affected boy with profound ID and a complex heart defect. The question rises whether this severe and atypical presentation, including respiratory insufficiency, duodenal stenosis, and full cheeks, can solely be attributed to the p.(Arg333Lys) MEIS2 variant. Interestingly, this variant involves the same highly conserved Arg333 residue, affected by an in-frame deletion in the patient reported by Louw et al. [10]. As this residue is involved in the multimeric contact of MEIS2 [27], variants in this domain might exert a dominant negative effect by interference with the assembly of the multimeric compound, hereby contributing to the more severe phenotype observed in these two patients. Unfortunately, patient 9 died at the age of 13 months, and no tissue was available to perform functional analyses. Alternatively, his atypical features could be ascribed to multilocus genomic variation [28], as additionally he was (1) compound heterozygous for two rare variants in LRP2, and (2) he had a de novo intragenic 185-bp deletion of FOXP1. The patient’s phenotype was not reminiscent of Donnai-Barrow (or faciooculoacousticorenal) syndrome [29] (OMIM:222448), which is caused by biallelic variants in LRP2. However, the FOXP1 deletion should be taken into account as loss-of-function variants and deletions of this gene are a known cause of global developmental delay, severe speech delay and dysmorphic features, including frontal bossing, downslanting palpebral fissures, and a short, broad nose [30]. Interestingly, two features in our patient, duodenal atresia [31] and complex CHD [32], were previously attributed to deletions or variants of FOXP1. However, it is important to note that FOXP1 is subjected to alternative splicing and that the 185-bp deletion is an intronic variant in the predominant expressed transcript and only affects an exon in a shorter alternative transcript. Therefore, the contributory effect of this FOXP1 deletion on the phenotype remains speculative. Finally, premature birth at 31 weeks and complications from multiple invasive procedures might have contributed to worse outcome (e.g., respiratory insufficiency, inguinal hernia, small for gestational age, bitemporal narrowing, nephrocalcinosis, and pancreatitis) in patient 9. His dizygotic twin brother has mild developmental delay without any major congenital anomalies.
Recurrent features of 15q14 deletions, harboring at least one exon of MEIS2, include palatal and heart defects and ID, which are ascribed to haploinsufficiency of this gene. For phenotypic delineation of the 15q14 deletion syndrome, we retained 25 deletion carriers from 24 independent families. Palatal defects ranged from bifid uvula to overt cleft palate, and were present in 18 out of 24 deletion patients (75%), as well as in all four members of a family with an inherited intragenic duplication in MEIS2. Congenital heart defects were reported in 12 out of 24 deletion patients (50%), and mainly were septal defects. Given the high prevalence of heart defects in the de novo MEIS2 variant cohort, haploinsufficiency of MEIS2 was considered a sufficient cause of CHD in the deletion cohort. Whether neighboring genes on 15q14 contributed to or independently caused the genesis of CHD in the deletion cohort remains a subject of debate. In 14 patients, the 15q14 deletion comprised ACTC1, a known cause of septal heart defects and of autosomal dominant cardiomyopathy. The prevalence of CHD tends to be higher in patients with 15q14 deletions including ACTC1 (70% (7/10), compared with those without ACTC1 (39% (5/13)), but was not different from the CHD prevalence in the cohort of patients with de novo MEIS2 variants (63%). None of the 14 patients with haploinsufficiency of ACTC1 were diagnosed with dilated or hypertrophic cardiomyopathy. It is currently unclear whether whole-gene deletions of ACTC1 predispose to cardiomyopathy. Nevertheless, surveillance guidelines for patients with 15q14 deletions, encompassing ACTC1, should include cardiologic follow-up, even in the absence of CHD.
Developmental outcomes were available for 17 deletion patients and ranged from learning problems to severe ID. Moderate-to-severe ID was more prevalent in patients with 15q14 deletions, affecting MEIS2 among other genes (10/17 (59%)), compared with patients with de novo variants [10, 11] or intragenic deletions/duplications [4, 6] in MEIS2 (4/14 (28%)), although no statistical significance could be reached (Fisher’s exact test p = 0.14). Deletions extending distally from MEIS2 (beyond the gene SPRED1), as well as deletions reaching proximally into 15q13.3 can be associated with poor developmental outcome. Microcephaly is another recurrent feature of 15q14 deletions, that is seen less commonly in the cohort with de novo MEIS2 variants (2/11 (18%) versus 8/17 (47%), Fisher’s exact test p = 0.22). Although we presume that haploinsufficiency of neighboring genes on 15q14 independently affect brain growth and neurocognitive development, we were unable to identify additional candidate genes for ID or microcephaly on 15q14 based on the current data. Café-au-lait (CAL) spots were reported in patients D, E, F, and L, and in family 4 by Johansson et al. [6]. These deletions extend distally from MEIS2 and affect SPRED1, the causal gene for Legius syndrome, which is characterized by multiple café-au-lait macules, intertriginous freckling, lipomas, and learning disabilities [33] (OMIM: 611431). CAL spots were not reported in patients G, K, and N, and the patients described by Chen et al. [3, 7] and by Brunetti-Pieri et al. [2], although the deletions in these patients encompass SPRED1. This could be attributed to young age at diagnosis, variable expressivity, or inaccurate phenotyping.
Although no recognizable facial gestalt could be ascribed to this syndrome, one frameshift variant was found by targeted sequencing of MEIS2 in a girl with a clinical suspicion of this syndrome. In addition to CHD and CP, this girl presented with thin, arched eyebrows, short alae nasi, and a thin vermillion, recurrent features in patients with deletions or loss-of-function variants of this gene (Fig. 1). In conclusion, deletions, indels or nucleotide variants in MEIS2 should be considered in every patient presenting with syndromic palatal defects, particularly if associated with CHD, developmental delay, and facial dysmorphism.
References
Erdogan F, Ullmann R, Chen W, et al. Characterization of a 5.3 Mb deletion in 15q14 by comparative genomic hybridization using a whole genome "tiling path" BAC array in a girl with heart defect, cleft palate, and developmental delay. Am J Med Genet A. 2007;143A:172–8.
Brunetti-Pierri N, Sahoo T, Frioux S, et al. 15q13q14 deletions: phenotypic characterization and molecular delineation by comparative genomic hybridization. Am J Med Genet A. 2008;146A:1933–41.
Chen CP, Lin SP, Tsai FJ, Chern SR, Lee CC, Wang W. A 5.6-Mb deletion in 15q14 in a boy with speech and language disorder, cleft palate, epilepsy, a ventricular septal defect, mental retardation and developmental delay. Eur J Med Genet. 2008;51:368–72.
Crowley MA, Conlin LK, Zackai EH, Deardorff MA, Thiel BD, Spinner NB. Further evidence for the possible role of MEIS2 in the development of cleft palate and cardiac septum. Am J Med Genet A. 2010;152A:1326–7.
Roberti MC, Surace C, Digilio MC, et al. Complex chromosome rearrangements related 15q14 microdeletion plays a relevant role in phenotype expression and delineates a novel recurrent syndrome. Orphanet J Rare Dis. 2011;6:17.
Johansson S, Berland S, Gradek GA, et al. Haploinsufficiency of MEIS2 is associated with orofacial clefting and learning disability. Am J Med Genet A. 2014;164A:1622–6.
Chen CP, Chen CY, Chern SR, et al. Prenatal diagnosis and molecular cytogenetic characterization of a de novo 4.858-Mb microdeletion in 15q14 associated with ACTC1 and MEIS2 haploinsufficiency and tetralogy of Fallot. Taiwan J Obstet Gynecol. 2016;55:270–4.
Shimojima K, Ondo Y, Okamoto N, Yamamoto T. A 15q14 microdeletion involving MEIS2 identified in a patient with autism spectrum disorder. Hum Genome Var. 2017;4:17029.
Gambin T, Yuan B, Bi W, et al. Identification of novel candidate disease genes from de novo exonic copy number variants. Genome Med. 2017;9:83.
Louw JJ, Corveleyn A, Jia Y, Hens G, Gewillig M, Devriendt K. MEIS2 involvement in cardiac development, cleft palate, and intellectual disability. Am J Med Genet A. 2015;167A:1142–6.
Fujita A, Isidor B, Piloquet H, et al. De novo MEIS2 mutation causes syndromic developmental delay with persistent gastro-esophageal reflux. J Hum Genet. 2016;61:835–8.
Larsen KB, Lutterodt MC, Laursen H, et al. Spatiotemporal distribution of PAX6 and MEIS2 expression and total cell numbers in the ganglionic eminence in the early developing human forebrain. Dev Neurosci. 2010;32:149–62.
Zerucha T, Prince VE. Cloning and developmental expression of a zebrafish meis2 homeobox gene. Mech Dev. 2001;102:247–50.
Melvin VS, Feng W, Hernandez-Lagunas L, Artinger KB, Williams T. A morpholino-based screen to identify novel genes involved in craniofacial morphogenesis. Dev Dyn. 2013;242:817–31.
Glickman NS, Yelon D. Cardiac development in zebrafish: coordination of form and function. Semin Cell Dev Biol. 2002;13:507–13.
Paige SL, Thomas S, Stoick-Cooper CL, et al. A temporal chromatin signature in human embryonic stem cells identifies regulators of cardiac development. Cell. 2012;151:221–32.
Machon O, Masek J, Machonova O, Krauss S, Kozmik Z. Meis2 is essential for cranial and cardiac neural crest development. BMC Dev Biol. 2015;15:40.
Kupchik GS, Revah-Politi A, Stong N, Anyane-Yeboa K. Expanding genotype and phenotype of MEIS2-related neurocristopathy (P11.070B). Presented at the European Society of Human Genetics 28 May 2017, Copenhagen, Denmark.
Wright CF, Fitzgerald TW, Jones WD, et al. Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data. Lancet. 2015;385:1305–14.
Homsy J, Zaidi S, Shen Y, et al. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science. 2015;350:1262–6.
Firth HV, Richards SM, Bevan AP, et al. DECIPHER: database of chromosomal imbalance and phenotype in humans using ensembl resources. Am J Hum Genet. 2009;84:524–33.
Ioannidis NM, Rothstein JH, Pejaver V, et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet. 2016;99:877–85.
Belcaro C, Dipresa S, Morini G, Pecile V, Skabar A, Fabretto A. CTNND2 deletion and intellectual disability. Gene. 2015;565:146–9.
Kumar R, Corbett MA, van Bon BW, et al. THOC2 mutations implicate mRNA-export pathway in X-linked intellectual disability. Am J Hum Genet. 2015;97:302–10.
Rigaud S, Fondaneche MC, Lambert N, et al. XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature. 2006;444:110–4.
Di Benedetto D, Musumeci SA, Avola E, et al. Definition of minimal duplicated region encompassing the XIAP and STAG2 genes in the Xq25 microduplication syndrome. Am J Med Genet A. 2014;164A:1923–30.
Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–97.
Posey JE, Harel T, Liu P, et al. Resolution of Disease Phenotypes Resulting from Multilocus Genomic Variation. N Engl J Med. 2017;376:21–31.
Donnai D, Barrow M. Diaphragmatic hernia, exomphalos, absent corpus callosum, hypertelorism, myopia, and sensorineural deafness: a newly recognized autosomal recessive disorder? Am J Med Genet. 1993;47:679–82.
Le Fevre AK, Taylor S, Malek NH, et al. FOXP1 mutations cause intellectual disability and a recognizable phenotype. Am J Med Genet A. 2013;161A:3166–75.
Hamdan FF, Daoud H, Rochefort D, et al. De novo mutations in FOXP1 in cases with intellectual disability, autism, and language impairment. Am J Hum Genet. 2010;87:671–8.
Chang SW, Mislankar M, Misra C, et al. Genetic abnormalities in FOXP1 are associated with congenital heart defects. Hum Mutat. 2013;34:1226–30.
Brems H, Chmara M, Sahbatou M, et al. Germline loss-of-function mutations in SPRED1 cause a neurofibromatosis 1-like phenotype. Nat Genet. 2007;39:1120–6.
Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1.
Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4.
Acknowledgements
We thank the patients and their parents for their cooperation. J.B. is funded by a clinical research fund of the University Hospitals Leuven (Klinisch onderzoeksfond KOF Leuven). This study makes use of data generated by the DECIPHER community. A full list of centers who contributed to the generation of the data is available from https://decipher.sanger.ac.uk/ and via email from decipher@sanger.ac.uk. Funding for the project was provided by the Wellcome Trust. The DDD study presents independent research commissioned by the Health Innovation Challenge Fund [grant number HICF-1009-003], a parallel funding partnership between the Wellcome Trust and the Department of Health, and the Wellcome Trust Sanger Institute (grant number WT098051). The views expressed in this publication are those of the author(s) and not necessarily those of the Wellcome Trust or the Department of Health. The study has UK Research Ethics Committee approval (10/H0305/83, granted by the Cambridge South REC, and GEN/284/12 granted by the Republic of Ireland REC). The research team acknowledges the support of the National Institute for Health Research, through the Comprehensive Clinical Research Network. This study was supported by New York State Genetics Services Program C029418. This work was supported by grants U01HL131003, UM1HL098147, UM1HL098123, UM1HL128761, UM1HL128711, and UM1HL098162 in support of the Pediatric Cardiac Genomics Consortium from the National Heart, Lung, and Blood Institute and the Eunice Kennedy Shriver National Institute of Child Health and Human Development. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Heart, Lung, and Blood Institute, Eunice Kennedy Shriver Institute of Child Health and Development, or the National Institutes of Health.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Verheije, R., Kupchik, G.S., Isidor, B. et al. Heterozygous loss-of-function variants of MEIS2 cause a triad of palatal defects, congenital heart defects, and intellectual disability. Eur J Hum Genet 27, 278–290 (2019). https://doi.org/10.1038/s41431-018-0281-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-018-0281-5
This article is cited by
-
Spatial enhancer activation influences inhibitory neuron identity during mouse embryonic development
Nature Neuroscience (2024)
-
The autism-associated Meis2 gene is necessary for cardiac baroreflex regulation in mice
Scientific Reports (2022)
-
MEIS2 (15q14) gene deletions in siblings with mild developmental phenotypes and bifid uvula: documentation of mosaicism in an unaffected parent
Molecular Cytogenetics (2021)
-
N6-methyladenosine dynamics in neurodevelopment and aging, and its potential role in Alzheimer’s disease
Genome Biology (2021)
-
Transcriptome analysis of neural progenitor cells derived from Lowe syndrome induced pluripotent stem cells: identification of candidate genes for the neurodevelopmental and eye manifestations
Journal of Neurodevelopmental Disorders (2020)
