
Abstract
Background
Galactosemia has not been recognized as a cause of extreme neonatal hyperbilirubinemia, although growing evidence supports this association.
Methods
In a retrospective cohort study, we identified children with galactosemia due to GALT deficiency using the Danish Metabolic Laboratory Database. Among these, we identified children with extreme neonatal hyperbilirubinemia or symptoms of ABE. Extreme neonatal hyperbilirubinemia was defined as maximum total serum bilirubin (TSBmax)) level ≥450 µmol/L and a ratio of conjugated serum bilirubin/TSB <0.30.
Results
We identified 21 children with galactosemia (incidence:1:48,000). Seven children developed extreme neonatal hyperbilirubinemia (median [range] TSBmax level: 491 [456–756] µmol/L), accounting for 1.7% of all extreme neonatal hyperbilirubinemia cases. During the first 10 days of life, hyperbilirubinemia was predominantly of unconjugated type. Four children developed symptoms of intermediate/advanced ABE. One additional child had symptoms of intermediate/advanced ABE without having extreme neonatal hyperbilirubinemia. On follow-up, one child had KSD.
Conclusions
Galactosemia is a potential cause of extreme neonatal hyperbilirubinemia, ABE, and KSD. It is crucial that putative galactosemic children are treated aggressively with phototherapy to prevent ABE and KSD. Thus it is important that galactosemia is part of the work up for unconjugated hyperbilirubinemia.
Similar content being viewed by others


Introduction
Hyperbilirubinemia is the most prevalent complication during the neonatal period.1 Usually, the unconjugated serum bilirubin level increases during the first days of life and declines toward the end of the first week. Occasionally, extreme neonatal hyperbilirubinemia develops. The native bilirubin is toxic and is capable of passing the blood–brain barrier due to its lipophilicity. If the level exceeds the neuroprotective defense mechanism, it can result in acute bilirubin encephalopathy (ABE) mainly caused by neuronal damage in the basal ganglia and in various brainstem nuclei.1, 2 ABE consists of three phases. During the early phase, the child is jaundiced, sucks poorly, lethargic, and hypotonic. The intermediate phase is characterized by weak Moro reflex, moderate stupor, fever, high-pitched cry, irritability, alternating hypotonia–hypertonia, retrocollis, and opisthotonus. The advanced phase is characterized by the absence of Moro reflex, coma, hypertonia, pronounced retrocollis and opisthotonus, high-pitched cry, apnea, no feeding, seizures, and occasionally subsequent death.3 The early phase is reversible, whereas children are at severe risk of developing chronic bilirubin encephalopathy (kernicterus spectrum disorder (KSD)) after having intermediate or advanced phases of ABE.1, 3 KSD is characterized by choreoathetotic/dystonic cerebral palsy, auditory dysfunction, mental retardation, paralysis of upward gaze, and enamel dysplasia.3 A distinct pattern on magnetic resonance imaging (MRI) scans and sensorineural hearing loss can be used to confirm the presence of KSD.2
Recent evidence suggests that galactosemia due to galactose-1-phosphate uridyltransferase (GALT) deficiency can cause extreme neonatal hyperbilirubinemia.4, 5 Galactosemia is an inherited, autosomal-recessive inborn error of galactose metabolism.6 It has an acute onset, often during the first week of life, the initial symptoms being poor feeding, vomiting, jaundice, lethargy, and hypotonia. Further, most children develop life-threatening complications, including hepatic failure, coagulopathy, and sepsis. Without rapid initiation of a galactose-free diet, galactosemia will run a fatal course in most children. However, despite galactose-free diet, long-term sequelae, including speech problems like verbal dyspraxia, psychomotor retardation, ataxia, tremor, and ovarian failure, develops.6
The aim of the study was to assess the incidence of children with galactosemia due to GALT deficiency in Denmark during a 16-year period (2000–2015) and to assess the frequency of extreme neonatal hyperbilirubinemia, ABE, and KSD in these children with galactosemia. Further, we investigated the incidence of extreme neonatal hyperbilirubinemia caused by galactosemia.
Methods
Study design
This is a nationwide retrospective descriptive cohort study in Denmark, covering a 16-year period from January 1, 2000 to December 31, 2015. The data were retrieved from both the Danish Metabolic Laboratory Database and the Danish Database of Extreme Neonatal Hyperbilirubinemia. The former database contains the personal identification numbers of all patients suffering from metabolic diseases, while the latter contains personal identification numbers of all children with extreme neonatal hyperbilirubinemia. The Danish Health Authority (3-3013-1624/1/) and the Danish Data Protection Agency (2008-58-0028,2016-67) approved the study.
Data retrieval
We identified all children diagnosed with galactosemia due to GALT deficiency using the Danish Metabolic Laboratory Database. We only included children with symptoms leading to a galactosemia diagnosis or children diagnosed prenatally due to older siblings having galactosemia. Further, we identified whether children had concomitantly extreme neonatal hyperbilirubinemia or ABE symptoms. These children were the main focus of our study.
Data were extracted by reviewing medical and laboratory records retrieved from pediatric departments throughout Denmark. We assessed the clinical course of the children to identify intermediate/advanced ABE or KSD. Intermediate/advanced ABE was defined as: high-pitched cry, opisthotonus, retrocollis, alternating hypertonia and hypotonia, hypertonia, lack of Moro reflex, or fever. KSD was defined as: choreoathetotic/dystonic cerebral palsy, auditory dysfunction, paralysis of upward gaze, or enamel dysplasia.
We defined extreme neonatal hyperbilirubinemia as a maximum total serum bilirubin (TSBmax) level ≥ 450 µmol/L combined with a conjugated serum bilirubin (CSB)/TSB ratio <0.30.7 Both levels were determined at the time of TSBmax or at the first fractionated blood sample afterwards.
During the study period, galactosemia was not included in the Danish nationwide neonatal screening program, thus suspicion of galactosemia is obtained by symptoms and laboratory test results. Presenting symptoms were defined as symptoms reported by the parents to the admitting physician, whereas symptoms at admission and symptoms developed during admission were defined as symptoms observed by health-care providers. Age at the start of galactose-free diet was defined as the age of the child at the day of galactose-free diet initiation, thus not necessarily the same day as breast milk or formula was discontinued.
Statistical analysis
Categorical variables were reported as counts with percentages; continuous variables as medians with ranges.
The incidence of galactosemia in Denmark was calculated from the total number of children diagnosed with galactosemia and the total number of live births. In addition, the frequency of all cases of extreme neonatal hyperbilirubinemia caused by galactosemia in relation to all cases of extreme neonatal hyperbilirubinemia was calculated using data from the Danish National Database of Extreme Neonatal Hyperbilirubinemia.
Results
Incidence of galactosemia and child characteristics
Of the 998,251 live-born children during the study period, 21 children were diagnosed with galactosemia in Denmark (incidence: 1:48,000).
Among the 21 children, the distribution of sex was equal (Table 1). All were full term, with a normal birth weight and prenatal period. The majority of the children were Northern European. Three children, diagnosed prenatally, received a galactose-free diet from birth, and had a normal neonatal period. Overall, p.Gln188Arg was the most frequent GALT gene mutation, and we found no correlation between genotype and extreme neonatal hyperbilirubinemia.
Extreme neonatal hyperbilirubinemia and ABE
Of the 18 children not diagnosed prenatally, 7 (39%) developed extreme neonatal hyperbilirubinemia. The frequency of extreme neonatal hyperbilirubinemia associated with galactosemia was 1.7%, as a total of 408 children developed extreme neonatal hyperbilirubinemia in Denmark during the study period. Among the 7 children with extreme neonatal hyperbilirubinemia, median [range] TSBmax level was 508 [456–756] µmol/L, and the children had a median [range] age of 6 [5–9] days at TSBmax level. Four of the seven children with extreme neonatal hyperbilirubinemia had weight loss ≥10%.
Of the seven children with galactosemia and extreme neonatal hyperbilirubinemia, four had symptoms compatible with intermediate/advanced ABE, with TSBmax levels between 456 and 756 µmol/L. Further, one additional child had symptoms compatible with intermediate/advanced ABE despite a TSBmax level of 374 µmol/L and a CSB/TSB ratio of 0.36, which, however, was measured 2 days after TSBmax. We found no competing conditions possibly leading to increased bilirubin toxicity or blood–brain barrier permeability in this child.
Among the above 8 children, median [range] age at symptom presentation was 4 [3–6] days. Often children were jaundiced and lethargic at admission, and during admission, they often developed vomiting and hepatomegaly. Symptoms of intermediate/advanced ABE, such as high-pitched cry, opisthotonus, retrocollis, hypertonia, lack of Moro reflex, and fever, were observed in the above-mentioned five children. No other reasons for these symptoms could be identified.
Regarding management of galactosemia, the 8 children had a median [range] age of 14 [9–67] days at the start of the galactose-free diet. Immediately following admission, all 8 children received intensive phototherapy for hyperbilirubinemia with either double phototherapy (n = 5) or triple phototherapy (n = 3), 1 was treated with albumin infusion, and none received exchange transfusion. Further, all children had normal hemoglobin levels on the day of admission (Table 1).
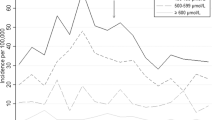
Figure 1 shows that, during the first 10 days of life, hyperbilirubinemia was predominantly due to unconjugated bilirubin. Conversely, after day 15 of life, bilirubin was predominantly conjugated.

Serum levels of total bilirubin and conjugated bilirubin in each galactosemic child with extreme neonatal hyperbilirubinemia or symptoms compatible with intermediate/advanced acute bilirubin encephalopathy (ABE). Case 1, 3, 4, 7, and 8 had symptoms compatible with intermediate/advanced ABE. Case 1 developed kernicterus spectrum disorder (KSD). Arrow represents age of the children at the start of galactose-free diet. For one child (case 8), represented by asterisk (*), galactose-free diet was initiated at age 67 days
Long-term follow-up
Of the five children with intermediate/advanced ABE, one had symptoms, including choreoathetotic/dystonic cerebral palsy, psychomotor retardation, speech retardation, fine tremor, and cataract, compatible with both KSD and long-term sequelae of galactosemia (Table 1). An MRI scan revealed no abnormalities at age 4 years. Owing to the mental retardation and cerebral palsy, the child could not cooperate with an auditory examination at age 8 years. Of the remaining four children with symptoms compatible with intermediate/advanced ABE, one had speech problems, which is consistent with long-term sequelae of galactosemia and three had normal development. These children did not have MRI scans done. Auditory examination was normal in two, not performed in one, and not feasible to do in the latter child because of persistent middle ear infections; however, the clinical impression was that hearing was normal. Finally, of the three children with no symptoms of ABE during the neonatal period, all had long-term sequelae of galactosemia. No enamel dysplasia was observed in any of the children.
A detailed description of each child’s clinical course is included in Supplementary Table S1–2 (online).
Discussion
To our knowledge, this is the first nationwide study reporting data on children with galactosemia with extreme neonatal hyperbilirubinemia. Our key findings were that 21 children had galactosemia, and of these, 8 developed extreme neonatal hyperbilirubinemia or symptoms of intermediate/advanced ABE. During the initial course of galactosemia, extreme neonatal hyperbilirubinemia was predominantly of unconjugated type, later changing to conjugated type. At follow-up, one child had KSD.
We found an incidence of galactosemia of 1:48,000 in Denmark during the 16-year study period. A previous study from Denmark reported an incidence of 1:85,000, which, however, only included one child in a 2-year period (2002–2004).8 The incidences of galactosemia in Ireland, Greece, Spain, South-West Germany, and Sweden are 1:16,000, 1:22,000, 1:43,000 1:78,000, 1:110,000, respectively.9,10,11,12,13 The varying incidences may be attributed to population differences and the sensitivity of the methods used to detect GALT deficiency.
Galactosemia is not listed among causes of extreme neonatal hyperbilirubinemia.14, 15 Thus it is surprising that 39% of all clinically presenting children with galactosemia in Denmark developed extreme neonatal hyperbilirubinemia accounting for 1.7% of all cases of extreme neonatal hyperbilirubinemia in Denmark. From a worldwide perspective, only 13 children with galactosemia and extreme neonatal hyperbilirubinemia have previously been reported.5, 7, 16,17,18,19,20,21,22,23,24 Thus, together with our findings of the 7 children with galactosemia and extreme neonatal hyperbilirubinemia, a total of 20 children have currently been reported. In studies from Australia, Canada, the Netherlands, the UK and Ireland, and Switzerland regarding extreme neonatal hyperbilirubinemia, no cases associated with galactosemia were reported among a total of 555 children.25,26,27,28,29 Interestingly, an overrepresentation of children from Denmark has been observed. This may be attributable to the general understanding that galactosemia mainly causes hyperbilirubinemia of the conjugated type. Possibly, this leads investigators to exclude children with galactosemia from extreme neonatal hyperbilirubinemia surveys.14, 15 Further, various definitions of extreme neonatal hyperbilirubinemia may be a contributing factor, as only children with a CSB/TSB ratio <0.20 were included in the study from the Netherlands.25
We observed that early in the neonatal period (i.e., <10 days from birth) extreme neonatal hyperbilirubinemia was predominantly of the unconjugated type. This is surprising as previous evidence suggests that hyperbilirubinemia associated with galactosemia is mainly of the conjugated type.14, 15 Possibly, the predominance of unconjugated serum bilirubin observed in our study may be caused by hemolysis due to galactose-1-phosphate accumulation in erythrocytes, added to the low activity of uridine 5′-diphospho-glucuronosyltransferase in the liver, and increased enterohepatic recirculation of bilirubin due to reduced food ingestion.4, 14, 30 In our study, the hemoglobin levels were normal at admission and the considerable weight loss, partly caused by dehydration, cannot explain the lack of low hemoglobin concentrations. In addition, low levels of hemoglobin are only seen at severe hemolysis.
We used a rather high CSB/TSB ratio to define extreme neonatal hyperbilirubinemia in our study, as measurement of fractionated TSB often overestimates CSB.7 CSB/TSB ratios were often not available until a few days after TSBmax level potentially meaning that actual CSB/TSB ratios were even lower than those reported. This was the case in the ABE symptomatic child, who did not have extreme neonatal hyperbilirubinemia, where the CSB/TSB ratio was high but not measured until 2 days after TSBmax level. However, giving the highly characteristic symptoms, we concluded that the child probably had intermediate/advanced ABE. In line with this, cases of intermediate/advanced ABE have been reported in children with TSBmax levels in the range 340–425 µmol/L.18, 31 We found no other competing conditions potentially leading to increased bilirubin toxicity, increased blood–brain barrier permeability, or explaining the symptoms. As previously stated, galactosemia runs a fatal course in most children if galactose-free diet is not initiated rapidly. However, in the child without extreme neonatal hyperbilirubinemia, galactose-free diet was not initiated until day 67. Here it is possible that galactosemia did not run a fatal course, because the child most of the time received intravenous fluid treatment. This may further explain the diagnostic delay of galactosemia.
Among the 13 children with galactosemia and extreme neonatal hyperbilirubinemia in previously published case reports and studies, six developed intermediate/advanced ABE.5, 7, 16, 18, 19, 24 Clearly, it is crucial that clinicians are aware of galactosemia as a possible etiology in children with extreme neonatal hyperbilirubinemia to prevent development of ABE. As such, galactosemia should potentially be part of the diagnostic work up of unconjugated hyperbilirubinemia. This is especially relevant considering the prompt response to initiation of a galactose-free diet with rapid reduction of hemolysis and liver involvement and consequently hyperbilirubinemia.17 Accordingly, we advise clinicians to initiate galactose-free diet at suspicion of galactosemia. In our study, children received intensive phototherapy immediately following admission, and as observed by others, this resulted in a drastic decline in TSBmax.32 Therefore, no children received exchange transfusion.
Concurrent with our findings, intermediate/advanced phases of ABE can be reversible, thus not necessarily leading to long-term sequelae.16 In our study, only one of the five children with symptoms of intermediate/advanced ABE had signs of KSD. Among the 13 published cases of extreme neonatal hyperbilirubinemia associated with galactosemia, three cases of KSD have been described,5, 18, 19 only one of whom had MRI findings consistent with the condition.5 Consequently, we would like to stress that galactosemia can lead to extreme neonatal hyperbilirubinemia of such impact that KSD may develop.
The strength of our study was the use of nationwide cohorts, whereas the small number of patients with galactosemia caused by the relative rarity of the disease is a limitation.
Conclusions
Galactosemia may be a cause of extreme neonatal hyperbilirubinemia, ABE, and KSD. Consequently, aggressive phototherapy should be considered even in children with putative galactosemia experiencing borderline hyperbilirubinemia. Galactosemia should potentially be part of the diagnostic work up in children with unconjugated hyperbilirubinemia and included in guidelines as well. Currently, this is being implemented in Danish nationwide guidelines.
References
Bhutani, V. K., Vilms, R. J. & Hamerman-Johnson, L. Universal bilirubin screening for severe neonatal hyperbilirubinemia. J. Perinatol. 30, S6–S15 (2010).
Shapiro, S. M. Chronic bilirubin encephalopathy: diagnosis and outcome. Semin. Fetal Neonatal Med. 15, 157–163 (2010).
American Academy of Pediatrics Subcommittee on Hyperbilirubinemia. Management of hyperbilirubinemia in the newborn infant 35 or more weeks of gestation. Pediatrics 114, 297–316 (2004).
Woo, H. C., Phornphutkul, C. & Laptook, A. R. Early and severe indirect hyperbilirubinemia as a manifestation of galactosemia. J. Perinatol. 30, 295–297 (2010).
Sahoo, T. et al. Galactosaemia: an unusual cause of chronic bilirubin encephalopathy. BMJ Case Rep. 2015, bcr2014206852 (2015).
Bosch, A. M. Classical galactosaemia revisited. J. Inherit. Metab. Dis. 29, 516–525 (2006).
Ebbesen, F. Recurrence of kernicterus in term and near-term infants in Denmark. Acta Paediatr. 89, 1213–1217 (2007).
Lund, A. M. et al. Biochemical screening of 504,049 newborns in Denmark, the Faroe Islands and Greenland--experience and development of a routine program for expanded newborn screening. Mol. Genet. Metab. 107, 281–293 (2012).
Coss, K. P. et al. Classical Galactosaemia in Ireland: incidence, complications and outcomes of treatment. J. Inherit. Metab. Dis. 36, 21–27 (2013).
Schulpis, K. et al. Screening for galactosaemia in Greece. Paediatr. Perinat. Epidemiol. 11, 436–440 (1997).
Varela-Lema, L. et al. Appropriateness of newborn screening for classic galactosaemia: a systematic review. J. Inherit. Metab. Dis. 39, 633–649 (2016).
Lindner, M. et al. Efficacy and outcome of expanded newborn screening for metabolic diseases--report of 10 years from South-West Germany. Orphanet. J. Rare. Dis. 6, 44 (2011).
Ohlsson, A., Guthenberg, C. & von Dobeln, U. Galactosemia screening with low false-positive recall rate: the Swedish experience. JIMD Rep. 2, 113–117 (2012).
Holton, J. B., Walter, J. H. & Tyfield, L. A. in The Metabolic and Molecular Bases of Inherited Disease (eds Scriver CR, Beaudet AL, Sly WS, Valle D) 1553–1583 (McGraw Hill, New York, NY, 2001).
Piazza, A., & Stoll, B. in Nelson Textbook of Pediatrics (eds. Kliegman R.) 756–761 (Elsevier/Saunders, Philadelphia, PA, 2011).
Hansen, T. W. et al. Reversibility of acute intermediate phase bilirubin encephalopathy. Acta Paediatr. 98, 1689–1694 (2009).
Honeyman, M. M. et al. Galactosaemia: results of the British Paediatric Surveillance Unit Study, 1988-90. Arch. Dis. Child. 69, 339–341 (1993).
Johnson, L. et al. Clinical report from the pilot USA Kernicterus Registry (1992 to 2004). J. Perinatol. 29, S25–S45 (2009).
Berry, G. T. Galactosemia: when is it a newborn screening emergency? Mol. Genet. Metab. 106, 7–11 (2012).
Cheng, S. W., Chiu, Y. W. & Weng, Y. H. Etiological analyses of marked neonatal hyperbilirubinemia in a single institution in Taiwan. Chang Gung Med. J. 35, 148–154 (2012).
Levy, H. L., Pueschel, S. M. & Hubbell, J. P. Jr. Unconjugated hyperbilirubinemia in galactosemia. N. Engl. J. Med. 292, 923–924 (1975).
Choy, Y. S. et al. A new variant of classical galactosemia due to a novel mutation in the GALT gene. J. Inherit. Metab. Dis. 35, S73 (2012).
Christensen, R. D. et al. Unexplained extreme hyperbilirubinemia among neonates in a multihospital healthcare system. Blood Cells Mol. Dis. 50, 105–109 (2013).
AlOtaibi, S. F., Blaser, S. & MacGregor, D. L. Neurological complications of kernicterus. Can. J. Neurol. Sci. 32, 311–315 (2005).
Gotink, M. J. et al. Severe neonatal hyperbilirubinemia in the Netherlands. Neonatology 104, 137–142 (2013).
Manning, D. et al. Prospective surveillance study of severe hyperbilirubinaemia in the newborn in the UK and Ireland. Arch. Dis. Child. Fetal Neonatal Ed. 92, F342–F346 (2007).
Zoubir, S. et al. Incidence of severe hyperbilirubinaemia in Switzerland: a nationwide population-based prospective study. Arch. Dis. Child. Fetal Neonatal Ed. 96, F310–F311 (2011).
Sgro, M., Campbell, D. & Shah, V. Incidence and causes of severe neonatal hyperbilirubinemia in Canada. CMAJ 175, 587–590 (2006).
McGillivray, A. et al. Prospective surveillance of extreme neonatal hyperbilirubinemia in Australia. J. Pediatr. 168, 82–87.e83 (2016).
Burton, B. K. Inborn errors of metabolism in infancy: a guide to diagnosis. Pediatrics 102, E69 (1998).
McGillivray, A. & Evans, N. Severe neonatal jaundice: is it a rare event in Australia? J. Paediatr. Child Health 48, 801–807 (2012).
Hansen, T. W. Acute management of extreme neonatal jaundice--the potential benefits of intensified phototherapy and interruption of enterohepatic bilirubin circulation. Acta Paediatr. 86, 843–846 (1997).
Author information
Authors and Affiliations
Contributions
L.F.B. conceptualized and designed the study, collected and interpreted data, drafted the initial manuscript, and reviewed and revised the manuscript. M.L.D., A.M.L., and F.E. conceptualized and designed the study and reviewed and revised the manuscript. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Bech, L.F., Donneborg, M.L., Lund, A.M. et al. Extreme neonatal hyperbilirubinemia, acute bilirubin encephalopathy, and kernicterus spectrum disorder in children with galactosemia. Pediatr Res 84, 228–232 (2018). https://doi.org/10.1038/s41390-018-0066-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41390-018-0066-0
This article is cited by
-
Bilirubin Induces A1-Like Reactivity of Astrocyte
Neurochemical Research (2023)
-
Management of severe hyperbilirubinemia in the cholestatic neonate: a review and an approach
Journal of Perinatology (2022)
