
Abstract
Sleep disturbances including insomnia independently contribute to risk of inflammatory disorders and major depressive disorder. This review and overview provides an integrated understanding of the reciprocal relationships between sleep and the innate immune system and considers the role of sleep in the nocturnal regulation of the inflammatory biology dynamics; the impact of insomnia complaints, extremes of sleep duration, and experimental sleep deprivation on genomic, cellular, and systemic markers of inflammation; and the influence of sleep complaints and insomnia on inflammaging and molecular processes of cellular aging. Clinical implications of this research include discussion of the contribution of sleep disturbance to depression and especially inflammation-related depressive symptoms. Reciprocal action of inflammatory mediators on the homeostatic regulation of sleep continuity and sleep macrostructure, and the potential of interventions that target insomnia to reverse inflammation, are also reviewed. Together, interactions between sleep and inflammatory biology mechanisms underscore the implications of sleep disturbance for inflammatory disease risk, and provide a map to guide the development of treatments that modulate inflammation, improve sleep, and promote sleep health.
Similar content being viewed by others
INTRODUCTION
Over one-quarter of the population of the United States experience sleep problems, and nearly a third of adults experience at least one insomnia complaint (Ohayon, 2002). In addition, insomnia is one of the most ubiquitous complaints in psychiatric populations, including adults with major depressive disorder (Irwin, 2015) and is reported in 50–70% of primary care patients (Shochat et al, 1999; Wittchen et al, 2001, Tubtimtes et al, 2009). Increasing evidence has demonstrated over the past decade that insomnia is not only a health complaint but also independently contributes to risk of infectious and inflammatory disease including depression, as well as all-cause mortality (Irwin, 2015). Understanding the biological mechanisms that underlie the risk between sleep disturbance such as insomnia and adverse health outcomes will refine identification of those at greatest risk to be targeted by early intervention and prevention of morbidity.
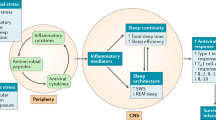
In this review, an integrated understanding of ‘sleep health’ (Buysse, 2014) is provided with a focus on the immune system. Specifically, this review examines the links between sleep and the innate immune system, given substantial epidemiological evidence that sleep disturbance (ie, insomnia, poor sleep quality, and/or insufficient sleep) contributes to inflammatory disease risk, and that sleep disturbance and inflammation are both thought to have a role in depression (Irwin, 2015). Further, we discuss the homeostatic role of sleep in the regulation of inflammatory biology dynamics, and how sleep disturbance and extremes of sleep duration impact the innate immune system at the systemic, cellular, and genomic levels with implications for inflammaging and molecular processes of aging (Figure 1). Sleep is also reciprocally linked to the innate immune system; substantial evidence from translational animal models has revealed that inflammatory cytokines homeostatically regulate sleep. Together, these findings support the potential to develop interventions that either target inflammation to improve sleep macrostructure, or to treat sleep complaints to reverse inflammation, with implications for promoting sleep health.

Sleep disturbance and innate immunity. Following a night of sleep loss, or during a period of sleep disturbance, nerve fibers from the SNS release the neurotransmitter norepinephrine into primary and secondary lymphoid organs and stimulate the adrenal gland to release stored epinephrine into the systemic circulation. Both neuromediators stimulate leukocyte adrenergic receptors (eg, ADRB2) and activate nuclear factor (NF)-κB-mediated inflammatory programs. Intrinsic circuits detect microbes via pattern recognition receptors (PRRs) such as the toll-like receptor 4 (TLR4) and stimulate inflammatory gene expression via transcription factors such as nuclear factor (NF)-κB. The production of proinflammatory cytokines interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α) occurs. Bidirectional links between the brain and periphery allow the brain to regulate inflammatory activity, and inflammatory activity in turn can influence neural processes in the brain and alter sleep. When this dynamic is induced by sustained sleep disturbance, a feed-forward dysregulation of sleep can occur, which may also confer activation of the conserved transcriptional response to adversity (CTRA). CTRA activation leads to increases in proinflammatory gene expression and increased risk for inflammation-related disorders such as cardiovascular disease, cancer, and major depressive disorder, and to decreases in antiviral gene expression and increased risk of infectious diseases.
SLEEP: CHARACTERISTICS AND ASSESSMENT METHODS
Sleep Characteristics
Arousal states follow a continuum from fully awake to deep sleep (ie, highly synchronous electroencephalography, EEG, and high arousal thresholds). Sleep consists of two major ‘phases’, rapid eye movement (REM) sleep and non-rapid eye movement (NREM) sleep. In humans, the phase of NREM sleep is further subdivided into four ‘stages’: stages 1–4, or more recently defined as three stages, N1, N2, and N3, which parallel a continuum of sleep depth. The transition from wakefulness to sleep occurs during Stage 1 sleep; sleep onset is defined by the occurrence of Stage 2 sleep and its two EEG characteristic features, spindles and K-complexes, superimposed on a background of low voltage EEG. Human Stages 3 and 4 sleep are referred to as slow-wave sleep (SWS) due to the preponderance of high-amplitude low-frequency components characteristic of the EEG during this stage; recent scoring methods combine stages 3 and 4 into a single category or N3 sleep.
The scoring of the EEG into stages of sleep follows standardized, but arbitrary criteria (Rechtschaffen and Kales, 1968), which should not be construed as meaning that sleep consists of discrete units that are quantal in nature. Rather, continuous progression from wakefulness to NREM sleep and REM sleep typically occurs. After NREM sleep, there is a transition to REM sleep; and after a period of REM sleep, there may or may not be a brief arousal or awakening. This cycle repeats throughout the night, with humans showing four to six NREM–REM cycles of ~80–110 min duration; rats will have many NREM–REM cycles, each lasting ~8–10 min. Furthermore, the interval between NREM and REM sleep is not stable across the night but rather becomes shorter over the course of successive cycles, in which SWS is preponderant in the first half of the night and REM sleep is preponderant in the second half of the night.
The distribution of sleep across the diurnal period can be either ‘monophasic’ or ‘polyphasic’. When sleep occurs during a single period of the 24-h cycle, as generally is the case with humans, this is a monophasic sleep pattern. In contrast, laboratory rodents (ie, rats and mice) show polyphasic sleep during both light and dark periods of a 24-h light:dark cycle, which is also strongly influenced by circadian factors. Hence, rats and mice sleep more during the light period of a light:dark cycle than they do during the dark period.
Although sleep was historically viewed as an organismal or whole-brain process, there is now ample evidence that some brain regions engage in sleep processes, while others simultaneously exhibit waking activity. The idea of ‘local sleep’ evolved from the hypothesis that local use-dependent activation of brain regions during wakefulness affects the EEG of those brain regions during subsequent sleep. This hypothesis was first proposed by Krueger in 1993 (Krueger and Obal, 1993), and then experimentally tested by Kattler, Dijk and Borbély (Kattler et al, 1994). Local sleep theories are now well-accepted and evidenced, in part by imaging studies that demonstrate links between regional EEG slow-frequency activity (see later) and brain metabolic activity in the same region during subsequent wakefulness (Wilckens et al, 2016). As such, the historical nature of sleep as a whole-brain phenomenon is now challenged by a new paradigm of local sleep, which has important ramifications for assessment of pathologies associated with sleep.
EEG Assessment of Sleep
To fully evaluate the substrates of sleep, it is necessary to determine sleep using EEG criteria with additional assessment of muscle tone (electromyogram (EMG)) and eye movements (electrooculogram (EOG)), important for staging the onset of REM sleep. Furthermore, multiple surface electrodes record the EEG over several regions of the cerebral cortex, and simultaneous recording of the EEG and either the EMG or body movements, allows determination of phases of sleep or wakefulness; the amplitude of the EMG, eg, diminishes from awake to sleep. The presence of random eye movements as assessed by the EOG, and low muscle tone as assessed by the EMG, are characteristic of REM sleep. Measures of sleep continuity include total sleep time, sleep latency, sleep efficiency (ie, percentage of time awake during the sleep period), and wake after sleep onset. As noted above, sleep architecture includes amounts of NREM sleep, including stages 1 (N1), stage 2 (N2) sleep and stages 3–4 (N3 or SWS) sleep. REM sleep includes measures of REM sleep amount, REM latency, REM density, and REM duration.
To further refine measurement of the amplitude and/or frequency components of the EEG waveform, spectral analyses are used. Spectral analyses are mathematical tools that are used to deconstruct complex EEG waveforms into constituent frequency components. Fast Fourier transformation is one type of spectral analysis that is commonly used in clinical sleep medicine and preclinical research. Fast Fourier transformation of the EEG yields units of power for select frequency bands. Frequency bands that are routinely reported in sleep studies include slow frequencies (<5 Hz, referred to as ‘delta’ frequency) and faster frequency bands, such as ‘theta’ (generally defined as 6–9 Hz) or ‘beta’ (12–14 Hz, spindle frequency activity). Power in these frequency bands is associated with NREM sleep (delta and spindle frequency bands) and with REM sleep (theta frequency band). During transitions from wakefulness to NREM sleep, EEG amplitude increases, leading to a shift from mixed EEG frequencies to predominately lower EEG frequencies (<5 Hz), or delta frequency band. In contrast, during the transition from NREM sleep to REM sleep, there is a reduction in the amplitude of the EEG relative to that during NREM sleep with an EEG shift from the low-frequency delta band to a higher frequency theta band that is accompanied by spindle activity. Further during REM sleep, EMG activity is reduced to its lowest level and there is an absence of generalized activity. Importantly, spectral analysis is a sensitive tool to evaluate the influence of such factors as age, medication use, and insomnia complaints on the EEG during sleep, which can further delineate discrepancies between subjective and objective measures of sleep.
There is a homeostatic drive to sleep; the longer we are awake, the sleepier we become (Borbely and Achermann, 1999). When prolonged wakefulness (sleep deprivation) is imposed, humans and animals sleep longer during the recovery night, yet do not fully recover all the sleep lost. The recovery night is also characterized by deeper sleep, which can be indexed by power in the delta frequency band during NREM sleep (sometimes referred to as delta power, or slow-wave activity). Whereas sleep lost during prolonged wakefulness may in part be functionally recovered by sleeping longer and with deeper intensity, some populations (ie, older adults, depressed patients, and abstinent alcohol-dependent subjects) evidence a defect in the homeostatic regulation of sleep, and fail to show the typical rebound increase in deep or SWS (Reynolds et al, 1987; Irwin et al, 2002). In contrast, REM sleep usually recovers more than was lost.
Actigraphic Assessment of Sleep
Polysomnography provides a laboratory-based measure of sleep and its macrostructure in humans. However, such laboratory assessment of sleep is not fully representative of sleep patterns in the community, and cannot be used to estimate disturbances of sleep, which might be occurring over days to weeks in a naturalistic setting. Hence, as research efforts increasingly focus on sleep health in community samples, sleep actigraphy is being used. The watch-shaped actigraph estimates sleep patterns and circadian rhythms, and when coupled with a sleep diary that notes when sleep onset and morning awakening occurs, can provide an assessment of sleep continuity. For example, high levels of activity are taken as a measure of wakefulness and low levels of activity are taken as a measure of sleep. Whereas these actigraphic estimates of sleep continuity are reliable and correlate with measures of total sleep time, sleep efficiency, and wake after sleep onset as characterized by polysomnography (Ancoli-Israel et al, 2003), actigraphy measures cannot replace the precision of polysomnography in the objective assessment of sleep. Nevertheless, in contrast to polysomnography, actigraphy provides an assessment of sleep in a naturalistic setting for weeks or possibly months at a time.
Behavioral Assessment of Sleep
Multiple brief self-report instruments have been validated to screen for insomnia complaints and assess symptom severity. Among the questionnaires that are easy to use is the seven-item Insomnia Severity Index, which assesses sleep quality, fatigue, psychological symptoms, and quality of life, with a high sensitivity and specificity for the detection of insomnia cases (Morin et al, 2011). Another questionnaire that is widely used and validated in various populations is the Pittsburgh Sleep Quality Index (Buysse et al, 1989; Cole et al, 2006). This 19-item self-report questionnaire evaluates seven clinically derived domains of sleep difficulties (ie, quality, latency, duration, habitual efficiency, sleep disturbances, use of sleeping medications, and daytime dysfunction), in which a threshold score shows high sensitivity and specificity for identifying clinically significant sleep impairment (Cole and Motivala et al, 2006). However, in many large-scale epidemiological or naturalistic field studies, it is possible to obtain only one or more questions about sleep complaints (ie, sleep quality and sleep duration). Even with constraints of comprehensive sleep assessment in large-scale studies, it is increasingly recognized that both assessment of sleep quality and sleep duration is necessary to capture two critical dimensions of sleep disturbance (Vgontzas et al, 2013), and these aspects of sleep together appear to be most strongly correlated with changes in inflammatory outcomes (Irwin et al, 2016). Further, the strength and quality of assessment of sleep disturbance (ie, validated questionnaire vs single item) contribute to heterogeneity of effects linking sleep to immune outcomes; stronger effects are found when questionnaires are used (Irwin and Olmstead et al, 2016). Yet, when use of validated questionnaires is not possible, it is important to note that certain single items predict daytime consequences, a necessary criterion for the diagnosis of insomnia (Kierlin et al, 2012). These predictive items show the following hierarchical order: self-reported dissatisfaction with sleep, complaints of non-restorative sleep, difficulty resuming or maintaining sleep, and difficulty initiating sleep (Ohayon et al, 2012).
Finally, the patient’s own sleep perceptions are critical for the diagnosis of insomnia. Indeed, polysomnography provides little information to confirm or exclude insomnia in a study of patients with insomnia and controls, with similar findings found in a large multicenter field trial (for a review, see (Vgontzas and Fernandez-Mendoza et al, 2013)). Indeed, the diagnosis of insomnia disorder in the Fifth edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-5) is based solely on the subjective complaints of difficulty initiating or maintaining sleep, early awakening, interrupted or non-restorative sleep, and associated impairments in daytime functioning, which must be present at least 3 nights per week and last for 3 months or longer (American Psychiatric Association and DSM-5 Task Force, 2013). Furthermore, insomnia is no longer identified simply as a complaint but is defined as Insomnia Disorder by both DSM-5 and the International Classification of Sleep Disorders-3. In recognition that insomnia is common in other disorders such as depression, DSM-5 classification acknowledges this comorbidity without assigning casual attribution, which are otherwise inherent in primary and secondary labels. The American Academy of Sleep Medicine does not recommend polysomnography for assessment of insomnia, although it is typically used in the evaluation of other sleep disorders such as sleep apnea (Littner et al, 2003). Insomnia complaints with short sleep duration (<5 h) may represent a more severe phenotype of insomnia disorder (Vgontzas and Fernandez-Mendoza et al, 2013).
INFLAMMATORY BIOLOGY DYNAMICS OF INNATE IMMUNITY
Innate immunity serves as the body’s first line of defense against tissue damage and microbial infection (Medzhitov, 2008). Monocytes, macrophages, and dendritic cells comprise the immune cell types of the innate immune system, and these cells circulate in the body using invariant receptors to detect a wide variety of pathogens. Within minutes to hours after recognition of a foreign challenge, these cells become activated, which initiates a cascade of inflammatory processes that help contain an infection and ultimately promote healing and recovery (Medzhitov, 2008).
Microbes or pathogen-associated molecular patterns are recognized by ‘hard-wired’ or highly conserved receptors of innate immune cells. This strategy of pattern recognition means that innate immune responses rely on receptors expressed on a relatively small number of immune cell types to detect and generate a response to a wide range of microbial diversity. When these pattern recognition receptors (PRRs) are activated, increase in inflammatory activity occur both locally (ie, at the site of tissue injury or infection) and systemically (Medzhitov, 2008).
Toll-like receptors (TLRs), found on macrophages, neutrophils, and dendritic cells, represent one class of PRRs (Medzhitov, 2008). In general, TLRs recognize conserved components of microbes including bacteria, viruses, and fungi. However, within the family or class of TLRs, there is some specificity of ligand recognition. For example, endotoxin or lipopolysaccharide (LPS), a major component of the outer membrane of Gram-negative bacteria, binds to TLR4 (Medzhitov, 2008), which initiates a conserved signaling cascade that is characterized by activation of key intracellular transcription factors such as nuclear factor-κB (NF-κB) and activator protein 1 (AP-1) (Karin, 2006). When NF-κB is activated, transcription of proinflammatory immune response genes such as Tnfα and Il1 occurs, which leads to translation and production of proinflammatory cytokines that serve to coordinate the inflammatory response (Karin, 2006). Hence, intracellular processes that occur at the level of the genome most proximally regulate the inflammatory response. Below we describe how sleep has an important role in the dynamics of innate immune responses including the effects of sleep disturbance to upregulate innate immune responses, as indexed by increases in cellular and genomic markers of inflammation.
INNATE IMMUNE SYSTEM DYNAMICS DURING NOCTURNAL SLEEP: CIRCADIAN INFLUENCES
During nocturnal sleep, both circadian- and sleep-dependent processes contribute to the regulation of inflammatory processes, and understanding changes in immune function observed during nocturnal sleep requires consideration of the influence of both sleep and circadian oscillators. To achieve this experimental aim and strictly separate the influences of sleep and circadian oscillators on immune function, comparison of the immune parameters of interest is required between a regular sleep–wake cycle and 24-hours of continuous wakefulness. Furthermore, such studies must control for the effects of social zeitgebers, maintain constant conditions during these two periods, and select participants who are following a regular sleep–wake activity schedule. Few such studies have been performed, especially those that have obtained multiple measurements across the entire 24-hour period to determine circadian rhythms. Nevertheless, several studies have overcome these methodological obstacles and found variations across a regular 24-hour period, although the contribution of sleep and circadian oscillators has not been dissociated for some of these variations.
For circulating concentrations of interleukin (IL)-6, eg, there are two peaks, at 1900 hours and again at 0500 hours, and these peaks appear to be driven by circadian processes (Vgontzas et al, 1999). Yet, when disturbances of sleep are imposed on this circadian rhythm (eg, administration of early-night sleep deprivation), the nocturnal increase of IL-6 is delayed (Redwine et al, 2000), with additional findings that total night sleep deprivation diminishes the increase in IL-6 by about half (Vgontzas and Papanicolaou et al, 1999). Hence, in the absence of sleep during the early part of the night, circulating levels of IL-6 remain low and relatively unchanged. However, even during this time of sleep deprivation, a transient peak in IL-6 occurs at 0100 hours indicating that circadian factors continue to influence IL-6 levels (Redwine and Hauger et al, 2000), and Dimitrov et al (2006) have found that monocyte production of IL-6 also peaks at 0200 hours even in the midst of sleep deprivation. Other data indicate that the chronic disruption of circadian rhythm magnifies inflammatory responses to challenge with endotoxin in an animal model of shift work or chronic jet lag (Castanon-Cervantes et al, 2010), along with an increase in inflammatory gene expression profiles in adipose tissue (Herrero et al, 2015) possibly due to disruption in the circadian clock gene Rev-erbalpha (Sato et al, 2014).
Such dynamic regulation of IL-6 during nocturnal sleep may have reciprocal effects on the central nervous system (CNS), and regulate neural functions. Along with nocturnal increase in IL-6, there is a robust increase in circulating levels of IL-6R (Dimitrov and Lange et al, 2006). Because neural tissues lack membane-bound IL-6 receptor (IL-6R), integrated action of IL-6 on brain and non-immune tissues requires the release of IL-6R to activate these tissues. Interestingly, the increase in IL-6R is most pronounced during the late part of the night (Dimitrov and Lange et al, 2006), consistent with evidence that IL-6 levels are prominently increased during the late part of the night, along with relative increase in REM sleep amounts (Redwine and Hauger et al, 2000).
In contrast to IL-6, nocturnal increase in tumor necrosis factor-α (TNF) and its soluble receptor appear to be driven primarily by circadian factors (Born et al, 1997), although there is evidence that sleep, but not circadian factors, is associated with TNF. During sleep, serum levels of TNF levels decrease. However, there is evidence of a striking night-time increase in the ability of monocytes to respond to challenge (ie, TLR-4 activation with LPS) (Dimitrov et al, 2015), and the enhancing effect of nocturnal sleep on stimulated monocytic TNF production adds to the notion that nocturnal sleep favors immune defense to a microbial challenge (Besedovsky et al, 2012).
The mechanisms that contribute to nocturnal increase in proinflammatory cytokines are not known. Some have speculated that danger signals such as reactive oxygen species, nucleotides (eg, adenosine triphosphate), and heat-shock proteins accumulate during the wake period, and then act like classical immunological stimulants leading to increased production of proinflammatory cytokines, which then support the initiation of adaptive immune responses (Besedovsky et al, 2012). Alternatively, proinflammatory cytokines peak during the early SWS-dominated portion of sleep, and SWS sleep is associated with the release of growth hormone (GH) and prolactin. Both of these neuroendocrine hormones are known to enhance the proliferation and differentiation of T cells as well as to promote type 1 cytokine activity (Lange et al, 2006; Besedovsky et al, 2012).
As discussed elsewhere, substantial evidence demonstrates a reciprocal relationship between the CNS and innate immunity (Dantzer et al, 2008, 2012; Irwin and Cole, 2011). Hence, such neural-immune signaling suggests the possibility of a homeostatic feedback loop between sleep and cellular inflammation. Bjurström et al (2016) recently examined cellular inflammation before and after a sleep period in patients with rheumatoid arthritis (RA) and controls, and found countervailing associations between proinflammatory cytokine production, sleep maintenance, and sleep depth (Figure 2). For example, higher levels of TLR-4 stimulated monocytic production of TNF at 2300 hours were associated with decrease in awake time after sleep onset and with increase in sleep efficiency. In turn, each of these sleep maintenance measures had an opposing relationship on TNF production at 0800 hours. In addition, higher levels of spontaneous- and stimulated production of IL-6 at 2300 hours were associated with increase in stage 3 and SWS, with evidence that stage 4 was associated with decreases in IL-6 production at 0800 hours (Figure 2). Together, these data support the hypothesis of a homeostatic relationship between sleep and cellular inflammation that is cytokine specific.

Associations between sleep efficiency and evening (a) and morning (b) levels of stimulated monocytic expression of TNF in RA patients and controls, as illustrated by estimated regression lines by group, controlling for depressive symptoms and physical health functioning. Curvilinear brackets indicate residual standard error. For stimulated production of TNF, results are presented as % of total number of monocytes expressing TNF, respectively, natural log-transformed.
SLEEP DISTURBANCE AND REGULATION OF INNATE IMMUNITY
Experimental and naturalistic studies in humans that have examined the associations between sleep and measures of innate immunity are constrained by several limitations. First, measures of innate immunity have been focused almost exclusively on levels of inflammatory markers or on the ability of immune cells in the circulation to be activated ex vivo. It is not known whether sleep alters innate immunity in other compartments of the body, including the brain. Second, the vast majority of studies have obtained only one or two measures of inflammation at a single time point, and the temporal dynamics of expression of various cytokines has not been mapped. Thirdly, many studies examine circulating markers of proinflammatory cytokines, or the expression of inflammatory cytokines in cells after in vitro stimulation, but not both. There are differences between levels of circulating cytokines and the extent to which cells can respond to stimulation. For example, if systemic levels of inflammation are already elevated, the ability of immune cells to become further activated may be attenuated; hence, blunted stimulated production of proinflammatory cytokines cannot be assumed to mean that systemic inflammation is low. Finally, there has been a focus on inflammatory cytokines without consideration of levels of anti-inflammatory cytokines (ie, IL-10), which together need to be assessed to capture the balance of the inflammatory state in vivo.
Experimental Sleep Deprivation and Circulating Markers of Inflammation
Experimental manipulation of sleep duration provides a robust evaluation of the role of sleep in the regulation of innate immunity and markers of inflammation. Indeed a variety of experimental approaches including total night sleep deprivation, partial night sleep deprivation (ie, awake time during the early- or late part of the night), or chronic sleep restriction (ie, reduction of sleep duration by 1 or more hours for multiple nights) have been used to test whether changes in sleep duration alter markers of inflammation. Partial night sleep deprivation and chronic sleep restriction are thought to mimic disturbances of sleep duration, which are ubiquitous in clinical populations such as depressed patients; hence, these experimental studies have translational implications in understanding the pathways by which inflammatory disease risk is elevated in persons with chronic sleep disturbance (Irwin, 2015). Interestingly, sleep fragmentation has not been systematically used as an experimental approach, even though loss of sleep efficiency and increase in wake after sleep onset are particularly prevalent in depressed patients and in older adults (Dew et al, 1996).
Among the first observations linking sleep deprivation to innate immunity was the evidence that prolonged sleep loss or 40 h of wakefulness induced elevated levels of IL-1-like and IL-2-like activity, and that this increase was independent of the cortisol circadian rhythm (Moldofsky et al, 1989). With the advent of cytokine-specific measures, as opposed to nonspecific bioassays, several additional studies have employed total night sleep deprivation. For example, as the ‘dose’ of sleep loss progressively increases over 4 nights, there is evidence of cumulative increase of C-reactive protein (CRP) (Meier-Ewert et al, 2004). In contrast, increase of TNF emerges as early as 1–2 nights (Chennaoui et al, 2011). Elevated levels of IL-6 do not occur until after 4 nights (Shearer et al, 2001); interestingly a 2 h nap appears to be protective and reverses elevated levels of IL-6 following 4 nights of sleep restriction (Vgontzas et al, 2007). Alterations in systemic markers of inflammation are not limited to proinflammatory cytokines. For example, a single night of sleep loss induces an activation of vascular endothelial markers (ie, E-selectin, s-intercellular adhesion molecule, s-ICAM-1)(Frey et al, 2007; Sauvet et al, 2010). However, it is not clear whether sleep deprivation increases the cumulative (ie, 24 h) levels of inflammation; Vgontzas et al (1999) found that sleep deprivation shifted the temporal pattern of circadian IL-6 secretion, with lower levels of IL-6 during the night and higher levels during the day.
Similar findings are found for partial night sleep deprivation. When repeated for several nights as chronic sleep restriction, 10 nights of partial sleep deprivation (eg, loss of 4 h of sleep per night) led to increased levels of CRP and IL-6 (Meier-Ewert et al, 2004; Haack et al, 2007). Moreover, there is evidence that even shorter periods of sleep restriction (ie, 7 nights) induce increase in plasma concentrations of IL-6 in men and women, increase of TNF in men only (Vgontzas et al, 2004), and increase in inflammatory transcripts of IL-1β, IL-6, and IL-17 (van Leeuwen et al, 2009), which persists even after a night of recovery sleep (van Leeuwen et al, 2009). Nevertheless, when sleep restriction or sleep fragmentation is limited to only 1 or 2 nights (Stamatakis and Punjabi, 2010; Schmid et al, 2011; Abedelmalek et al, 2013), or interrupted by daytime naps (Faraut et al, 2011) (Shearer et al, 2001), circulating levels of inflammatory markers do not appear to change (Irwin et al, 2015). Interestingly, among those with chronic sleep disturbance, there appears to be an increased vulnerability to inflammation following a bout of sleep loss. For example, in abstinent alcohol-dependent patients who report ongoing disturbances of sleep continuity, a single night of sleep loss triggers increase in IL-6 and TNF, which is not found in those who have no underlying sleep problems (Irwin et al, 2004).
In sum, experimental sleep deprivation has yielded mixed results due in part to differences in duration of sleep deprivation and the selection of the cytokine measure of inflammation (Irwin and Olmstead et al, 2016). Further, there is considerable variability in effect sizes (ES) between the various studies with many studies relying on small samples. Indeed, our recent meta-analysis did not find any significant effect of sleep deprivation, inclusive of total-, partial, and sleep restriction, on markers of CRP, IL-6, and TNF in healthy adults (Irwin et al, 2016), which stand in sharp contrast with findings that have evaluated upstream pathways of cellular and genomic markers of inflammation. As noted below, these studies have demonstrated that acute sleep loss, of even a limited 4-h duration, induces an activation of inflammatory signaling. Together, these data suggest that more persistent disturbances of sleep might be necessary for inflammatory signaling to be translated into subsequent increase in systemic markers of inflammation.
Experimental Sleep Deprivation and Cellular Dynamics and Transcriptome Profiles
Elevated circulating levels of proinflammatory cytokines may be a consequence of activation of monocytic populations, the primary immune source of IL-6 and TNF. However, there are other non-immune cellular sources of inflammatory cytokines, such as adipocytes or skeletal muscle, which may contribute to increase in systemic inflammation after sleep loss and might be activated by sleep loss due to adrenergic signaling (Irwin, 2015). Hence, further studies have interrogated specific immune cell populations and examined, eg, whether sleep loss leads to aberrant increase in the cellular production of proinflammatory as indexed by resting, ex vivo expression of inflammatory cytokines in monocytes, or by cellular production of IL-6 and TNF following stimulation of the TLR-4 receptor (Irwin et al, 2006, 2010). Such cellular production of IL-6 and TNF has been linked to inflammatory diseases such as RA (Andreakos et al, 2004) and heart failure (Satoh et al, 2005).
Among the first studies, Irwin et al (2006) demonstrated that a single night of partial night sleep loss induced a robust increase in the ability of monocytes to respond to ligation of TLR-4 with LPS (Figure 3). Subsequent research demonstrated that this response was more robust in females, as compared with males, and that females showed sustained elevations in production of proinflammatory cytokines throughout the day following sleep loss. Furthermore, induction of an inflammatory response appears to vary across the life span. Carroll et al (2015) found that age modulated the effects of partial sleep deprivation, such that younger adults had an increase in inflammatory cytokine production that was not present in older adults (Carroll et al, 2015). Such age-related differential effects suggest that sleep loss might contribute to risk of inflammatory disorders in younger adults by increasing cellular inflammation, whereas in older adults, sleep loss might increase the risk of infectious disease due to blunted TLR activation.

Representative expression of IL-6 and TNF in LPS-stimulated CD14+ cells from a participant at baseline and at PSD. Numbers indicate percentage of the fraction of CD14+ cells that are positive for TNF alone (upper left), TNF and IL-6 (upper right), and IL-6 alone (lower right). In the baseline condition, 72.1% of the CD14+ cells are negative for both IL-6 and TNF, whereas only 34.5% of the CD14+ cells are negative for both IL-6 and TNF in the PSD condition.
The resting or constitutive expression of proinflammatory cytokines by monocytes is also increased in response to sleep loss. Indeed, even in the absence of exogenous activation of innate immunity, such as LPS, and baseline production of IL-6, and TNF by monocytes is increased by sleep restriction, which suggests that sleep loss serves to prime monocyte populations leading to greater increase in the production of inflammatory cytokines in response to challenge (Irwin et al, 2015). Given that monocytes are a primary source for inflammatory cytokines in the peripheral blood, spontaneous production of proinflammatory cytokines may in turn induce increase in circulating inflammatory markers with consequent systemic inflammation and metabolic changes. Small elevations in circulating inflammatory mediators, eg, have been associated with type II diabetes mellitus, independent of adiposity (Festa et al, 2000) as well with cardiovascular disease (Ridker et al, 2003; Ridker and Cook, 2004).
Transcriptional pathways are also activated following a night of restricted sleep, with evidence of increaes in transcription of IL-6 and TNF (Irwin and Wang et al, 2006). Such increases in transcription appear to be due to an activation of nuclear factor (NF)-κB, the key transcription control pathway in the inflammatory signaling cascade (Irwin et al, 2008). Again, these responses were more marked in females as compared with males, which raises the possibility that targeting sleep disturbance in women may have the benefit of differentially reducing the risk of inflammation and possibly inflammatory disease including subtypes of depressive disorders.
To provide an understanding about the broader role of sleep in the regulation of inflammatory mechanisms, recent research has examined the impact of sleep loss on the signal transducer and activator of transcription (STAT) protein family (Irwin et al, 2015). STAT proteins act as a key signaling cascade mediating cytokine receptor-derived signals, and activation of STAT members serves to transduce signals through the cytoplasm and to function as transcription factors in the nucleus (Miklossy et al, 2013). Among the six distinct STAT members (ie, STAT1, STAT2, STAT3, STAT4, STAT5, and STAT6) (Yu et al, 2009), STAT3 is thought to be a mediator of inflammation (Levy and Lee, 2002), as both IL-6 and TNF have been shown to activate STAT3; IL-6 mediates its effects through the STAT3 pathway; and STAT3 competes with the transcription factor NF-κB (Gao et al, 2012). STAT1 is linked to interferon (IFN) signaling, and STAT5 is linked to IL-2 (Shuai and Liu, 2003). Moreover, there is cross-regulation between STAT family members; STAT1 and STAT3 are both activated by IL-6 (Shuai and Liu, 2003). In addition, the specificity of IL-6 signaling is modulated by activation of STAT3 because removal of STAT3 results in prolonged STAT1 activation by IL-6. Finally, there is cross-talk between the two main cytokine signaling pathways, STATs and NF-κB (Shuai and Liu, 2003), in which activation of STAT3, eg, promotes NF-κB and IL-6 pathways, but opposes STAT1 and NF-κB-mediated T-helper (Th)1 immune response (Yu et al, 2009). Similar to the activation of other inflammatory mechanisms, partial night sleep deprivation was found to increase baseline, spontaneous monocytic expression of activated STAT1 and STAT5; this increase was delayed and found in the second morning after sleep loss indicating a sustained effect of sleep deprivation on STAT activation. The implications of increase in STAT5 are not fully understood, although constitutively active STAT5, as well as STAT3, is detected in numerous malignancies (Miklossy et al, 2013). In contrast, STAT1 contributes to antiviral immunity.
To further map the dynamics of sleep loss on the molecular signaling pathways that regulate inflammatory and other immune responses, the effects of sleep loss on transcriptome dynamics have been explored, with evidence that a single night of partial sleep loss induces an upregulation of a gene ensemble that includes the master circadian regulator, several immediate early genes marking cellular signal transduction, and multiple inflammatory response genes (Irwin et al, 2006). Among the transcription factor-binding motifs that were overrepresented in the sleep deprivation condition were promoters of genes involved in regulation by cyclic adenosine monophosphate/protein kinase A (cAMP/PKA)-induced transcription factors of the cAMP response element-binding protein/activating transcription factor (CREB/ATF) family, the protein kinase C-induced AP-1 family, the proinflammatory NF-κB/Rel family, and the mitogen-activated protein kinase-inducible E26 transformation-specific (ETS) transcription factor family typified by ELK1. Together, these experimental findings reveal that sleep regulates multiple signal transduction pathways including the NF-κB inflammatory signaling system.
Naturalistic Sleep Disturbance and Systemic Markers of Inflammation
(Figure 4) A variety of studies using epidemiological and naturalistic cross-sectional designs have evaluated the associations between sleep disturbance (insomnia complaints, poor sleep quality, and/or insufficient sleep) and inflammation in community- and clinical populations, with varying findings (Irwin, 2015, 2016). Yet, differences in the characterization of sleep disturbance, varying assessment methods used to evaluate sleep disturbance (ie, sleep quality and sleep complaints) and sleep duration, and various markers of inflammation, have made it difficult to establish confident conclusions about the association between sleep disturbances and inflammation. Hence, we recently undertook a systematic evaluation of the associations between sleep disturbance and sleep duration on inflammatory outcomes, and related effect size (ES), to understand the magnitude and specificity of different aspects of sleep (ie, sleep disturbance and sleep duration) on inflammation. (Irwin et al, 2016) A total of 72 studies (n>50 000) were analyzed with assessment of CRP, IL-6, and TNF. Studies evaluating sleep disturbance were categorized into three groups as determined by the assessment method: insomnia symptom reporting (single or multiple items); questionnaire; or diagnosis. Studies evaluating sleep duration were grouped into those that treated sleep duration as a continuous measure subjectively or objectively versus those that categorized sleep duration as short or long sleep, with the reference category for sleep duration set at 7–8 h per night. Finally, for sleep duration, the assessment method was considered, ie, self-report or objective.

Effects of sleep disturbance and extremes of sleep duration on cellular and molecular processes of inflammation including activation of NF-κB, inflammatory gene expression, production of proinflammatory cytokines, and increase in systemic inflammation as indexed by C-reactive protein (CRP).
This meta-analysis provided the first global evidence linking sleep disturbance, sleep duration, and inflammation in adult humans and found the following results with a high statistical power conferred by nearly 34 000 participants for CRP and over 3000 participants for IL-6. Sleep disturbance was associated with higher levels of CRP (ES.12; 95% CI=0.05-0.19) (Figure 5) and IL-6 (ES.20; 95% CI=0.08–0.31) (Irwin et al, 2016). Shorter sleep duration, but not the extreme category of short sleep, was associated with higher levels of CRP (ES.09; 95% CI=0.01–0.17) but not IL-6 (ES.03; 95% CI: −0.09 to 0.14). The extreme category of long sleep duration was associated with higher levels of CRP (ES.17; 95% CI=0.01–0.34) and IL-6 (ES.11; 95% CI=0.02–20). Neither sleep disturbances nor sleep duration was associated with TNF, although this conclusion is tempered by low statistical power with only 672 participants. Finally, it is not surprising that the ES linking sleep disturbance with IL-6 were larger than those found for CRP. It is known that IL-6 induces CRP; hence, sleep disturbance might have proximal effects on IL-6 and increase of CRP is found only when sleep is more persistent or more severe.

Forest plot of sleep disturbance associated with inflammation as indexed by C-reactive protein. Sleep disturbance is assessed by self-reported symptoms and questionnaires. Results are expressed as ES and 95% CI.
The ES linking sleep disturbance to inflammation are comparable, and larger in some cases, than the effects of some demographic and other biobehavioral factors on inflammation. For example, Liukkonen et al (2007) showed that elevated levels of CRP found in association with sleep disturbance (ie, self-reported problems sleeping) were greater than elevated levels associated with sedentary physical activity, low socioeconomic status, and tobacco smoking, but not with elevated body mass index. Likewise, Christian (Christian et al, 2011) found that self-reported poor sleep quality was correlated with CRP and IL-6 at a magnitude comparable to that between age and these markers of inflammation, but less than that found between BMI and CRP or IL-6; these latter findings are similar to those reported by Friedman et al (2005) Alternatively, the absolute change in levels of CRP or IL-6 with a healthy diet intervention (Neale et al, 2016), or aerobic exercise (Hayashino et al, 2014), is comparable to the absolute difference found in association with sleep disturbance.
Some heterogeneity among studies was found which could be due to differences in the assessment of sleep disturbance. For example, when sleep disturbance was assessed using more robust and validated measures, increase in CRP and IL-6 was found. Yet, when assessment was by simple symptom reporting with reliance on only a single question, there were mixed effects, suggesting that when quality of sleep assessment is stronger, an association with inflammation is more likely to be found.
Differences in sample characteristics might also contribute to heterogeneity across studies. Indeed, meta-regression results suggested that larger ES were associated with younger age and greater proportion of female subjects within the sample (Irwin et al, 2016). However, these sex differences were only statistically significant for sleep disturbance predicting IL-6 and sleep duration continuously predicting CRP. Nevertheless, given evidence that women appear to be especially vulnerable to the effects of sleep loss on cellular inflammation (as noted above) (Irwin et al, 2008, 2010), it is interesting to note that Suarez (2008) found that overall poor sleep quality was associated with increased inflammatory biomarkers, but only for women, similar to the findings of Friedman et al (2005) in older adults. Moreover, sex differences emerge in association with short sleep duration, as women, but not men, are more likely to show elevated levels of inflammation. For example, in the Whitehall II epidemiological study (n=4677) (Miller et al, 2009), women, but not men, who slept less than 8 h tended to have higher levels of IL-6 as well as higher levels of CRP. Moreover, in one of the few prospective studies of sleep disturbance and inflammation, Prather et al (2013) evaluated 980 participants with established cardiovascular disease, and followed 626 of these subjects for 5 years. Although subjective sleep quality was unrelated to inflammatory markers in cross-sectional and prospective analyses in the whole sample, poorer subjective sleep quality was prospectively associated with 5-year increase in IL-6, CRP, and fibrinogen in women but not men (Prather et al, 2013). Together, these findings parallel the risk profile for cardiovascular disease, in which subjective symptoms of disturbed sleep are associated with a greater risk in women than in men, even after control for relevant confounders (Irwin, 2015).
Other research suggests that quality of social ties might buffer the adverse effects of sleep disturbance on inflammation. Conversely, social isolation might exaggerate these associations. For example, poor sleep efficiency was found to be associated with higher levels of IL-6 (Friedman et al, 2005), especially in women with poor social relationships. Likewise in pre-hypertensive and hypertensive individuals, social support moderated the association between sleep disturbance and elevated levels of IL-6 and CRP, in which poor sleep appeared to confer a risk of increased inflammation only in those participants who also reported low social support (Tomfohr et al, 2015). Finally, in the CARDIA study, a population-based longitudinal study conducted in four US cities (n=2962 African-American and White adults), sleep disturbance (ie, insomnia complaints and short sleep duration) was a significant predictor of inflammation (CRP and IL-6) 5 years later (Cho et al, 2015) (Figure 6). Furthermore, subjective social isolation was a significant moderator of this association between sleep disturbance and inflammation in which this relationship was significantly stronger in those who reported feelings of social isolation. These findings that more precisely characterize the risk profiles linking sleep disturbance to inflammation have immediate implications for the development of more refined, clinical interventions that target sleep disturbances to reduce inflammation. Taken together, these data suggest that females and those who feel socially isolated may be at greatest risk and are most likely to benefit from prevention approaches (Cho et al, 2015).

Mean CRP level at follow-up according to baseline sleep disturbance score in socially integrated (n=1502) and socially isolated (n=1460) subgroups. Error bars represent SEM.
Multiple aspects of sleep behaviors are characterized when sleep disturbance is evaluated by questionnaire and diagnosis, and it is not known what aspect of sleep contributes to increase in inflammation. However, insomnia complaints when combined with short sleep duration are thought to be particularly caustic for health outcomes (Vgontzas et al, 2009, 2013; Irwin, 2015). In regards to inflammation, most studies have predominantly examined sleep disturbance and sleep duration in separate models. Sleep fragmentation is one insomnia complaint most associated with daytime dysfunction and report of poor sleep quality (Ohayon, 2005), yet systematic evaluation of sleep fragmentation has not been performed despite evidence that disruption of sleep continuity is uniquely associated with increased rates of mortality (Dew et al, 2003). Reports of sleep disturbance, as opposed to sleep duration, appear to carry unique risks, as Carroll et al (2015) found in a population-based sample from the Midlife Development in the United States survey (n=1023) in which self-reported poor sleep quality with either short or long sleep duration was associated with dysregulation in physiological set points across regulatory systems, as reflected in 22 biomarkers capturing cardiovascular, immune, lipid-metabolic, glucose-metabolic, sympathetic, parasympathetic, and hypothalamic–pituitary–adrenal systems (Figure 7).

Estimated mean and standard error of multisystem biological risk by sleep duration and PSQI global sleep score. Mean and standard error estimates derived from model after adjustments by age, gender, race, BMI, education, income poverty ratio, chronic conditions, and self-evaluated physical health. Multisystem Biological Risk score ranged from 0 to 7.
Meta-analytic findings have also examined extremes of sleep duration, and found that long sleep duration, but not short sleep duration, were associated with increase in CRP and with increase in IL-6 (Irwin et al, 2016). The absence of an effect of short sleep duration is surprising, although consistent with experimental data. Several high-quality studies have found no relationship between self-reported and objective (ie, polysomnographic) short sleep duration and markers of inflammation (Larkin et al, 2005; Taheri et al, 2007; Marsland et al, 2008; Lee et al, 2009; Rief et al, 2010; Ramey et al, 2012), even when measures of sleep quality and sleep efficiency were correlated with inflammation in the same population (Friedman et al, 2005). Nevertheless, several other large-scale studies have shown that short sleep duration, as well as long sleep duration, impact inflammation. In the population-based InCHIANTI study (n=751), levels of TNF, but not IL-6 or CRP, were increased at extremes of both short and long sleep as compared with those who slept 7–8 h (Stenholm et al, 2011), and Grandner et al (2013) found that elevated levels of CRP were found at extremes of sleep duration, but different patterns were observed for various ethnic groups, ie, non-Hispanic white (elevated CRP for<5 h and>9 h), black/African-American (elevated CRP for<5 h and 8 h), Hispanic/Latino (elevated CRP for>9 h), and Asian/ Other (higher in 9 and>9 h and lower in 5 and 6 h). Moreover, ethnoracial groups also demonstrated patterning by sex. To further complicate these observations, the effects of sleep duration might depend on the proinflammatory cytokine; Patel et al (2009) found that each additional hour of sleep duration resulted in an increase in CRP and an increase in IL-6, whereas a shorter sleep duration was associated with an increase in TNF. Given these heterogeneous findings, further examination of sleep duration, co-morbid insomnia complaints, and inflammation in relation to morbidity outcomes is needed to mechanistically extend epidemiological observations of an U-shaped association between sleep duration and mortality risk, in which long sleepers (>8 h per night) have a 30% greater risk, whereas short sleepers (<7 h per night) have a 12% greater risk of dying than those who sleep 7–8 h per night (Youngstedt and Kripke, 2004). To this end, (Smagula et al, 2016) objectively measured sleep characteristics using in home polysomnography and wrist actigraphy, along with assessment of CRP, IL-6, TNF, TNFrII, and IFN, in community-dwelling older men (n=2531) with follow-up on average for 7.4 years. Short sleep duration (<5 h) and sleep fragmentation (awake for⩾90 min during the night) was prospectively associated with increase in inflammatory burden, as defined by the number of inflammatory markers in the top quartile. The association between short (<5 h) sleep duration and mortality risk was attenuated to non-significance after adjusting for inflammatory burden or medical burden/lifestyle factors. In a separate sample of community-dwelling older adults, mortality risk was increased for those with short sleep duration (<6 h) and for those with long sleep duration (>8 h). Adjustment for levels of inflammation (CRP, IL-6, TNF) attenuated the mortality risk associated with short sleep duration, whereas the risk of long sleep duration was mainly attenuated by lifestyle and health factors.
SLEEP AND INFLAMMAGING: EXPERIMENTAL AND NATURALISTIC FINDINGS
Sleep loss activates inflammation and inflammatory signaling, and self-reported sleep disturbance (ie, insomnia complaints) is associated with increase in inflammation. Such elevated levels of inflammation are thought to contribute to biological aging. Indeed, ‘usual’ aging is associated with increased markers of inflammation or ‘inflammaging’(Franceschi and Campisi, 2014); proinflammatory secretory phenotypes and cellular senescence are critical elements in the related pathologies of aging including chronic diseases such as cancer, cardiovascular disease, dementia, arthritis, osteoporosis, sarcopenia, and immune compromise. Given that the prevalence of insomnia increases with age (Ohayon, 2002, 2004), and that insomnia complaints lead to inflammation (Irwin et al, 2015), there is much interest in understanding the impact of sleep on molecular aging.
The molecular causes of physical aging are thought to occur through the accumulation of cellular damage to mitochondrial DNA (mtDNA), telomeric ends, coding-DNA, and proteins (Kennedy et al, 2014). Together, damage to these multiple molecular components alters cellular energy production from the mitochondria, telomere length, inflammatory activity, and initiation of permanent cell cycle arrest (ie, cellular senescence) (Liu et al, 2013; Franceschi and Campisi, 2014). Cellular senescence, commonly reached by cell replication (eg, critically short telomeres) or excess cell stress (eg, DNA damage) (Blackburn, 2000, Campisi and d'Adda di Fagagna, 2007) is associated with the expression of p16(INK4a), a protein that inhibits cells from replicating (ie, a potent biomarker of human aging) (Liu et al, 2009). Cellular senescence is thought to further propagate nearby tissue aging through intracellular metabolic compromise (eg, mitochondrial dysfunction) (Sahin et al, 2011) and altered production of secretory factors that promote aging by increasing degradation of nearby tissue and sterile inflammation (Coppe et al, 2010a, 2010b; Freund et al, 2010) (ie, senescence-associated secretory phenotype, SASP). Through modifications to these basic molecular pathways involved in aging, mammalian aging can be delayed, raising the possibility that sleep disturbance and the development of interventional strategies that target sleep might mitigate age-related inflammatory sources and their systemic effect.
Given the potential mechanistic role of sleep as a behavioral factor that contributes to cellular aging, recent research has begun to examine the association between sleep disturbance (ie, insomnia complaints) and markers of cellular aging. For example, leukocytye telomere length is a biological marker of cellular aging, and Prather et al (2011) were among the first to examine associations of self-reported sleep duration, onset latency, and subjective quality with leukocyte telomere length in a community-dwelling women during midlife. Similar to the observations between sleep duration and inflammation, sleep duration was unrelated to leukocyte telomere length. However, poor sleep quality was associated with shorter leukocyte telomere length, independent of age, BMI, race, and income. Moreover, among those women for whom poor sleep quality was chronic, poorer sleep quality predicted shorter leukocyte telomere length. Additional work has extended these observations to older adults with assessment of sleep quality using a validated questionnaire (ie, Pittsburgh Sleep Quality Index, PSQI) and telomere length in peripheral blood mononuclear cells, and shown that age and self-reported sleep quality interact in predicting telomere length (Cribbet et al, 2014). Importantly, age was related to telomere length, and this association was more robust among poor sleepers. Moreover, among older adults with good sleep quality, the association between age and telomere length was attenuated (Cribbet et al, 2014). Carroll et al (2016) further examined this question in older adults and evaluated differences in telomere in relation to diagnostic insomnia, and determined whether insomnia interacts with chronological age to increase cellular aging. The results show that age groups (ie, 60–69 years versus 70–88 years) interact with insomnia diagnosis to predict shorter peripheral blood mononuclear cell telomere length. In the oldest age group (70–88 years), PBMC telomere length was significantly shorter in those with insomnia as compared with controls with no insomnia. However, in adults aged 60–69 years, PBMC telomere length was not different between insomnia cases and controls. Together these data suggest that clinically severe sleep disturbances (ie, insomnia) may increase cellular aging, especially in the later years of life.
To understand the causal role of sleep in inducing changes in molecular markers of cellular aging, Carroll et al (2016) has also examined whether partial night sleep deprivation would increase leukocyte gene expression of markers associated with cellular senescence, focusing on responses in older adults given prior findings that older adults are more likely to show telomere erosion in association with insomnia.(Figure 8) Following a single night of partial sleep loss, gene of the secretory-associated senescent phenotype was elevated, and increase was also found for NFKB2 and NFKB1, in which NFKB2 remained elevated even after a night of recovery sleep. Moreover, there was evidence that the sleep loss induced increase in genes associated with the composite DNA damage response (DDR), and this elevated expression remained elevated after recovery sleep. Finally, partial sleep deprivation altered genes involved in signaling senescence with increase in NBS1, CHK2, and ATM. Further, increase in the senescent signal marker p16INK4a (CDKN2A) was found to occur two nights after sleep loss. Together, sleep deprivation increases the DDR, increases senescence-associated secretory phenotypic expression pattern, and promotes pathways involved in the initiation of cellular senescence in aged adults. In sum, the DDR induced by sleep deprivation may be sufficient to signal growth arrest and drive senescence, especially in older adults who elevated levels of inflammatory signaling, which can be further increased by sleep loss. (Carroll et al, 2016).

Gene expression of the SASP and DDR at baseline, PSD, and 1 day after PSD (recovery). SASP is a composite score created from a sum of nine z-transformed genes. DDR is a composite score created from a sum of 30 z-transformed genes. Error bars represent standard error of estimated marginal mean, adjusting for BMI and sex. *p<0.05.
SLEEP DISTURBANCE AND DEPRESSION: ROLE OF INFLAMMATION
Sleep disturbance (ie, poor sleep quality) is an independent and robust predictor of depression risk (Irwin, 2015), yet the biological pathways that underlie this risk profile are not known. Given that sleep disturbance drives increase in inflammation, identification of inflammation as a pathway linking sleep and depression would have important clinical implications for development of strategies to prevent depression by targeting, eg, individuals with ‘two-hits’, sleep disturbance and inflammation (Cho et al, 2016). As extensively reviewed elsewhere, inflammation has a prominent role in depression (Dantzer, 2012; Raison and Miller, 2013), and multiple links exist between inflammation and depression (for a review, see (Slavich and Irwin, 2014). First, depression comorbidity is high in patients with an inflammatory disorder. Second, compared with non-depressed individuals, markers of inflammation are elevated in depressed patients. Furthermore, elevated levels of CRP and IL-6 prospectively predict the occurrence of depression (Gimeno et al, 2009). Third, acute physiological increase of inflammation causes increase in depressive symptoms. For example, experimental activation of inflammation activates brain sites that regulate positive and negative effects (Eisenberger et al, 2009, 2010a, 2010b), and is associated with increase in depressed mood, especially in women. Finally, antagonism of endogenous inflammation appears to reduce depressive symptoms, at least in those depressed patients who evidence an inflammatory subtype of depression with high level of inflammation (Tyring et al, 2006; Raison et al, 2013).
Symptoms of insomnia, including difficulties initiating and maintaining sleep, often co-occur with depression. However, when insomnia or persistent sleep disturbance occurs, it has a role in instigating depressive symptoms, and also predicts depression incidence (Ford and Kamerow, 1989). To date, more than 40 studies have tested the role of insomnia in the onset of depression (for a review, see (Riemann, 2010)) and meta-analytic findings show that sleep disturbance independently predicts a twofold increased risk of depression (Baglioni et al, 2011) or is one of the first clinical signs of a depressive disorder (Dryman and Eaton, 1991). In older adults who have had a prior history of depression but who are not depressed, persistent insomnia is particularly potent in increasing the risk of depression recurrence. Indeed, when insomnia complaints persist for up to one year, there is a 14-fold greater risk of depression in the year following as compared with those without sleep disturbance (Lee et al, 2013). Although some evidence suggests that this association appears to be specific to those with a history of depression (Cho et al, 2008), persistent insomnia was found to predict risk for depressive symptoms over 6 years even in those without a depression history (Jaussent et al, 2011).
To understand whether sleep disturbance might drive increase in inflammation that mediates the risk of depression, we have recently examined whether sleep disturbance acts as a vulnerability factor for depressed mood induced by an inflammatory challenge (ie, endotoxin administration) in healthy females vs males (Cho et al, 2016). Using an experimental model that mimics increased inflammation found in infections (Breen et al, 1990), autoimmune diseases (Ishihara and Hirano, 2002) and also psychological stress (Steptoe et al, 2007), we found that the presence of pre-existing sleep disturbance led to exaggerated increase in depressed mood response to inflammatory challenge as compared those who reported no sleep disturbance (Figure 9). Interestingly, specific sleep abnormalities appear to predict subsequent depression in response to inflammatory activation. Using quantitative EEG, Lotrich and co-worker (Lotrich and Germain, 2015) found that the delta sleep ratio (an index of early-night restorative delta power) was inversely associated with increase in depressive symptoms, as was elevated alpha power (8–12 Hz) in patients treated with IFN-alpha for hepatitis C. If low delta sleep ratio and high alpha power are indicators of vulnerability to depression, interventions to prevent depression may need to specifically target these specific sleep parameters.

Effect of endotoxin on depressed mood over time in females according to sleep disturbance. Depressed mood was assessed at baseline (T0) and then approximately every hour after injection for the next 6 h (T1–T6). T2 was assessed at 1 h and 40 min after injection; T3 was assessed at 3 h and 30 min after injection; and T4–T6 were assessed hourly after T3. Error bars indicate 95% confidence intervals. *Marginal means adjusted for age, race, BMI, and baseline depressive symptoms.
Consistent with evidence that inflammation induced depressed mood may be more robust in women as compared with men (Moieni et al, 2015), sleep disturbance (ie, poor sleep quality) is associated with exaggerated increase of depressive symptoms following inflammatory challenge mainly in females (Cho et al, 2016). Moreover, cytokine levels were more strongly correlated with depressed mood in females with sleep disturbance, despite similar overall increase in circulating proinflammatory cytokines in females with and without sleep disturbance, which suggest that sleep disturbance may increase the vulnerability to depression by augmenting affective sensitivity to cytokines and possibly by altering neural sensitivity to inflammation. The highly controlled experimental design of this study extends prior findings that have examined the complex inter-relationships between sleep disturbance, inflammation, and depression, which found that self-reported sleep disturbance predicted the onset of depression during interferon-α treatment (Franzen et al, 2010).
These clinical and experimental observations support a ‘two-hit’ model of depression, one that posits that sleep disturbance serves as a vulnerability factor and that subsequent exposure to heightened inflammatory states such as an infectious challenge or psychological stress triggers increase in depressive symptoms. Alternatively, we have found that experimental sleep disturbance induces greater increase in depressive symptoms in persons with an inflammatory disorder (Irwin et al, 2012), which suggests that inflammation might also serve as a vulnerability factor, in which subsequent exposure to sleep disturbance triggers increase in depressive symptoms (Cho et al, 2016).
MECHANISMS LINKING SLEEP AND IMMUNE SYSTEM DYNAMICS
Overview
As depicted in Figure 1, and described more fully by Slavich and Irwin, Slavich and Cole (Slavich and Cole, 2013; Slavich and Irwin, 2014) and Irwin and Cole (2011), the host defense program is activated by non-physical social, symbolic, anticipated, or imagined threats, with evidence that sleep disturbance leads to a similar response. The basal transcriptome profile associated with this response to adversity, including sleep loss, has been coined as the conserved transcriptional response to adversity (CTRA), and is characterized by increased proinflammatory and reduced antiviral skewing of the basal gene expression profile, which increases an individual’s risk for both viral infection and inflammation-related disease. (see also Irwin and Cole (2011); Antoni et al (2012); Fredrickson et al (2013)). For discussion about the effects of sleep on antiviral gene expression, adaptive immune responses, and infectious disease risk, see Irwin (2015).
Sleep influences two primary effector systems, the sympathetic nervous system (SNS) and hypothalamus–pituitary–adrenal axis, which together shift the basal gene expression profile toward an increased proinflammatory state. In relation to inflammation, the parasympathetic nervous system is also involved, with evidence that vagal activity can modulate immune responses at a regional level through both the efferent and afferent fibers of the vagus nerve, to prevent excessive inflammation (Sternberg, 2006; Tracey, 2009). The mechanisms underlying these pathways are described in detail elsewhere (Sternberg, 2006; Dantzer et al, 2008; Irwin and Cole, 2011). We only briefly summarize the pathways here, with an emphasis on how effector mechanisms in the SNS and HPA axis regulate inflammation. Further, we emphasize that the mechanisms that might explain the associations between sleep disturbance and inflammation are relatively unexplored.
Sympathetic Nervous System
Sleep disturbance can lead to activation of β-adrenergic signaling, which in turn induces increase in NF-κB, inflammatory gene expression, production of proinflammatory cytokines, and markers of systemic inflammation. (Figure 1) As described previously, the SNS allows the CNS to ‘steer’ innate immune responses between proinflammatory and antiviral phenotypes (Collado-Hidalgo et al, 2006; Cole, 2010; Irwin and Cole, 2011). By releasing the neurotransmitter norepinephrine into peripheral tissues, primary and secondary lymphoid organs, and all other major organ systems (including the vasculature and perivascular tissues), norepinephrine regulates immune response gene transcription and proinflammatory cytokine production via stimulation of β-adrenergic receptors (Nance and Sanders, 2007; Irwin and Cole, 2011), with some evidence suggesting that α-adrenergic signaling is also involved. Such adrenergic signaling suppresses transcription of antiviral type I interferon genes, as previously discussed elsewhere (Cole et al, 1998), and also upregulates transcription of the proinflammatory immune response genes IL1B, TNF, and IL6, leading to increase in systemic inflammatory activity (Cole, 2010).
The relevance of the SNS pathway to regulation of inflammation associated with sleep disturbance is several fold. Normal nocturnal sleep is associated with a drop in sympathetic outflow (Irwin et al, 1999). In the transition from wakefulness to sleep, a shift from sympathetic to parasympathetic outflow occurs, which is determined in part by the stage of sleep as well as sleep depth (Boudreau et al, 2013). For example, during NREM or SWS, there is a marked decrease in sympathetic activity, whereas REM sleep is associated with SNS outflow that is higher and similar to daytime levels (Somers et al, 1993). When sympathetic activity is characterized by plasma levels of the sympathetic neurotransmitter, a similar pattern is found (Irwin et al, 1999). Among patients with insomnia, increase in norepinephrine, epinephrine, and other markers of sympathetic outflow (as measured by blood pressure, heart rate variability, and impedance cardiography) occurs (Bonnet and Arand, 1997; Lanfranchi et al, 2009; Riemann, 2010; De Zambotti et al, 2011; Vgontzas and Fernandez-Mendoza et al, 2013). Further, it appears that sleep disturbance causes such sympathetic activation, as experimental sleep deprivation induces a similar increase in markers of sympathetic activity (Irwin and Thompson et al, 1999; Irwin and Ziegler, 2005). Interestingly, increase in urinary catecholamine and levels of their metabolites are more likely to occur in those insomnia patients who also report short sleep duration, a pattern similar to the greater activation of inflammatory biomarkers in this more severe phenotype (Vgontzas et al, 2013). However, to date, no research has simultaneously characterized whether insomnia-related increase in sympathetic outflow correlate, or potentially mediate, increase in inflammation, although experimental findings that manipulate perceived stress demonstrate that activation of sympathetic outflow mediates increase in inflammatory signaling.
Hypothalamic–Pituitary–Adrenal Axis
Activation of the HPA axis, the second effector pathway, leads to the release of one of the body’s most potent anti-inflammatory substances, the glucocorticoid cortisol, from the adrenal cortex. Under normal conditions, cortisol acts to suppress (rather than promote) transcription of proinflammatory immune response genes (Berkenbosch et al, 1989). The inhibitory effects of glucocorticoid activation on immune response gene transcription is mediated by at least three mechanisms. First, glucocorticoids can bind to receptors on gene promoter sequences, which interrupts proinflammatory gene expression. Second, certain anti-inflammatory genes are transcriptionally induced by glucocorticoid receptor (GR) activation, which in turn leads to inhibition of activation of the proinflammatory transcription factor NF-κB, thereby blocking the inflammatory cascade. Finally, proinflammatory transcription factors such as NF-κB and AP-1 can antagonize gene transcription via protein–protein interactions (Irwin and Cole, 2011). Together, these fundamental physiological mechanisms, a prototype for some of the most effective anti-inflammatory drugs, ensure that levels of inflammatory activity are regulated and that such glucocorticoid feedback inhibition of immune response gene transcription protects against diseases that involve excessive inflammation.
Although these dynamics characterize HPA axis functioning under normal conditions of typical, intermittent engagement, a different pattern can emerge that leads to HPA axis-related increase (as opposed to decreases) in inflammation (Stark et al, 2001, 2002; Miller et al, 2002; Avitsur et al, 2003). This process, referred to as ‘glucocorticoid resistance’, or ‘glucocorticoid insensitivity’, occurs when immune cells become less sensitive to the anti-inflammatory effects of glucocorticoids as a consequence of persistent or repeated activation of the HPA axis. Such glucocorticoid resistance may have adaptive significance, as cortisol provides the organism with the metabolic energy it needs to respond to threatening conspecifics; on the other hand, closely timed elevations in proinflammatory cytokines accelerate wound healing and limit infection if an injury occurs. It is not known whether persistent sleep disturbance can lead to glucocorticoid resistance, although these mechanisms have implications for depression. For example, individuals with major depressive disorder show flatter diurnal cortisol slopes (ie, higher overall cortisol concentrations) than in persons without depression, which is explained in part by varying levels of levels of glucocorticoid sensitivity or insensitivity (Pace et al, 2007; Jarcho et al, 2013). Given the role of sleep disturbance as a predictor of depression risk (Cho et al, 2008, 2016; Lee et al, 2013), possibly due to related increase in inflammation, research is needed to determine whether aberrant glucocorticoid signaling is a mechanism that can influence the risk of depression following persistent sleep disturbance.
In contrast to SNS, activity of the HPA system shows a reversed temporal pattern, with increase during the nocturnal period. During the early SWS-rich part of nocturnal sleep, release of hypothalamic corticotropin-releasing hormone (CRH), of pituitary corticotropin (ACTH) and of adrenal corticosteroids, reaches an absolute minimum; later during the night REM sleep-rich part of sleep, HPA secretory reaches a maximum at about the time of morning awakening. Both circadian and sleep-dependent mechanisms contribute to the regulation of HPA activity. For example, experimental stimulation of the HPA axis is blunted during times of SWS (Spath-Schwalbe et al, 1993; Bierwolf et al, 1997). Yet, circadian oscillators are robust, with evidence that sleep deprivation during a single night does not induce a substantial increase in cortisol release (Redwine et al, 2000).
The function of cortisol on the immune system in relation to sleep suggests that actions of this hormone are mediated not only via GR but also via mineralocorticoid receptors (MR). Binding affinity of cortisol in humans is about tenfold higher for MR than GR (de Kloet et al, 1994); hence, about 70–80% of MR are continuously occupied throughout the 24-hour cycle. In contrast, GR become predominantly occupied during periods of distinctly increased corticosteroid concentrations. During times of stress and around the maximum of the circadian oscillation, blockade of MR in blood from awake subjects (characterized by high cortisol concentrations) markedly enhances CD4+ and CD8+ T cells producing IL-2, IFN-γ, and TNFα, which are stronger than those of the selective GR blockade (with RU-486). Together, these data suggest that the suppressing effects of cortisol on T-cell-derived cytokines are to a substantial extent mediated via MR activation. When cortisol is added to blood sampled during SWS (characterized by minimum cortisol concentrations) a decrease in the percentage of CD4+ and CD8+ T cells producing IFN-γ, IL-2, and TNF is found. Suppression of endogenous cortisol release during the early night appears to drive facilitated production of type 1 cytokines, and possibly inflammatory cytokines. Prolactin and GH secreted during sleep also have a role in the regulation of the immune system, although their effects have predominantly been studied in relation to the production of type 1 cytokines and the adaptive immune responses as reviewed elsewhere (Besedovsky et al, 2012).
IMMUNE REGULATION OF SLEEP
Pathways of Communication Linking the Immune System and the Central Nervous System
Although once thought to be an immune-privileged site, it is now well-accepted that there is an innate immune system within the CNS. Communication from the peripheral immune system to the CNS occurs by several mechanisms, including direct neural innervation, actions of humoral mediators, and by active transport systems that move substances across the blood–brain barrier (BBB) (reviewed by Dantzer et al (2008)). Furthermore, the CNS is not a ‘passive recipient’ of information, but actively modulates aspects of peripheral immunity (reviewed by Olofsson et al (2012)). Thus, bidirectional communication exists between the CNS and the peripheral immune system that is actively driven in response to a variety of insults.
Similar to evolving concepts of CNS–immune interactions, changes in sleep were once thought to be merely a consequence or byproduct of the febrile response that accompanies many infections. We now know that sleep and fever may be experimentally dissociated during immune activation (eg, (Walter et al, 1989) and that changes in sleep during immune challenge is an actively driven process that facilitates the production of fever (Imeri and Opp, 2009) (Opp and Imeri, 1999). As such, data on sleep–immune interactions now demonstrate that these interactions are bidirectional: immune activation alters sleep, and disrupted sleep impacts immune function. A comprehensive review of mechanisms by which the CNS and peripheral immune system communicate is beyond the scope of this chapter. The interested reader is referred to some of the classic literature on this topic by Dantzer (eg, (Dantzer et al, 2008)).
Neural Mechanisms
Direct neural communication from the peripheral immune system to the CNS occurs by actions of cytokines and pathogen-associated molecular patterns (PAMPS) on the vagus nerve (reviewed by Dantzer et al (2008)). The vagus nerve diffusely innervates peripheral organs, and vagal afferents project to brainstem nuclei, including the nucleus of the solitary tract (NTS), the ventrolateral medulla (VLM), the paraventricular and supraoptic nuclei of the hypothalamus, and the amygdala. Each of these brain regions is implicated in the regulation and/or modulation of sleep. Furthermore, there are projections from the NTS to the parabrachial nucleus, and from the amygdala to the periaqueductal gray (PAG). The PAG and the VLM also project to the superficial dorsal horn of the spinal cord. As such, there are multiple afferent and efferent projection pathways that comprise circuits by which rapid CNS–peripheral immune system communication occurs.
Preclinical studies of neural communication between the CNS and the peripheral immune system have targeted the vagus nerve, and demonstrate that vagotomy (sub-diaphragmatic transection of the vagus nerve) blocks or alters the dynamics of peripheral immune system communication to the CNS (Bluthe et al, 1994; Laye et al, 1995; Bluthe et al, 1996). Depending on factors such as dose and/or timing, IL1- or LPS-induced alterations in sleep are blocked or attenuated in animals subjected to vagotomy (Hansen and Krueger, 1997; Opp and Toth, 1998; Zielinski et al, 2013). Neural communication from the peripheral immune system to the CNS is a critical component of responses to systemic infection because it is much quicker than the other forms of CNS–immune communication pathways and comprises the brain’s early warning system for immune activation.
Humoral Mechanisms
Effects of circulating immunomodulators on CNS processes have been extensively studied. TLRs are present on macrophage-like cells that are found in the circumventricular organs and the choroid plexus. When PAMPS activate TLRs in the circumventricular organs or the choroid plexus, these macrophage-like cells produce proinflammatory cytokines, which can enter the brain by volume diffusion (Vitkovic et al, 2000). In addition to actions of PAMPS on TLRs, IL1 receptors are present on endothelial cells, which when activated by circulating IL1 induce local prostaglandin E2 production (Quan et al, 1998), an inflammatory signal that triggers additional brain responses to immune activation.
Blood to Brain Transport Mechanisms
Active transporters on the BBB move an array of immunomodulatory molecules into and out of the CNS (reviewed by Banks (2015)). Active transport of substances across the BBB is controlled by the CNS, and differs in consequences from passive diffusion or unregulated leakage that is associated with BBB dysfunction. BBB active transport mechanisms are influenced by sleep and circadian processes (Pan and Kastin, 2016) and by sleep disruption (He et al, 2014). Within the context of sleep and CNS–immune interactions, the active transport of cytokines (Banks et al, 1991; Gutierrez et al, 1993; Plotkin et al, 1996), notably IL1, IL6, and TNF, is likely an important contributor to changes in sleep that occur during prolonged disease or infection. In addition to sleep disruption, BBB function also is altered in aging, which may explain, in part, why morbidity and mortality in oldest old individuals is higher than in young adults for chronic inflammatory diseases. For example, sleep disruption increases BBB transport of TNF in aged mice, but not in young mice (Opp and Krueger, 2015).
Animal Models
Early research effort focused on the impact of infection on sleep, which has been determined for many different microorganisms, including viral, bacterial, and fungal pathogens. Changes in sleep have also been determined for prion-related diseases and for protozoan parasites, but most studies to date have used virus and bacteria as the infectious agent.
CNS viral diseases, such as rabies (Gourmelon et al, 1991) or viral encephalitis in rodents after vesicular stomatitis virus infection (Machida et al, 2014), are associated with altered sleep. In these CNS infections, it is difficult to know whether altered sleep is due to direct actions on regulatory mechanisms, or whether it results from virus-induced brain lesions. One model that has been frequently used to determine effects of viral infections on sleep is influenza. Influenza virus localizes to the respiratory tract during the early stage of disease, and does not cause brain lesions. In addition, influenza infections pose tremendous public health burdens due to the millions of lives lost each year and the threat of pandemics. Smith and colleagues (Smith, 1992) report that low doses of influenza in humans increase sleep, and Drake et al (2000) demonstrated in healthy human volunteers that infection with rhinovirus 23 (HRV-23) increases total sleep time. (Rhinoviruses are the predominant cause of the ‘common cold’.) In rabbits, intravenous injections of influenza virus are also associated with large increase in NREM sleep and suppressed REM sleep, even though the virus does not replicate in this species (Toth, 1995).
Studies in mice infected with influenza virus demonstrate profound changes in sleep through the course of disease progression (Toth et al, 1994; Fang et al, 1995). As a preclinical model, influenza infection of mice is clinically relevant because mouse-adapted strains of this virus can be introduced into the respiratory tract and can fully replicate in the lungs, causing a severe acute phase response. Mice infected with influenza virus display profound increase in NREM sleep and inhibition of REM sleep, which last 3 or more days (Krueger et al, 1994). Macrophages appear to be the critical immune cell type driving increased NREM sleep, whereas NK cells, neutrophils, and T lymphocytes do not play a significant role (Toth and Hughes, 2004). There are strain differences in responses of mice to this challenge (Toth and Verhulst, 2003), indicating a genetic component affecting the sleep response to influenza virus (Trammell and Toth, 2008).
One generic viral component that increases NREM sleep is virus-associated double-stranded (ds) RNA. Virus-associated dsRNA induces numerous cytokines, including IL-1, IL-6, TNF, and IFN. Interferons have a major role in viral symptoms because influenza-infected IFN-receptor KO mice are less ill later in the infection and recover sooner (Traynor et al, 2007). Sleep modulatory cytokines, in addition to IFNs, likely mediate the sleep responses to influenza virus. For example, although the duration of altered NREM and REM sleep is the same in both strains after viral challenge, mice deficient in the 55 and 75 kD TNF receptors manifest reduced EEG delta power, whereas in wild-type control mice, delta power increases (Kapas et al, 2008). IL1 signaling in brain requires a brain-specific receptor accessory protein (Dinarello, 2009); mice lacking this accessory protein have higher morbidity and mortality, and they sleep less during the infection, than do wild-type mice.
Bacterial infections also modulate sleep. Studies of rabbits inoculated with the gram-positive bacteria Staphylococcus aureus were among the first to quantify changes in sleep through the course of an infectious process (Toth and Krueger, 1988). Within a few hours of S. aureus administration, NREM sleep is twice the amount as during comparable periods after control inoculation. This initial phase of increased duration and intensity of NREM sleep is followed by a more prolonged phase of decreased NREM sleep (Toth and Krueger, 1988). During both phases of the NREM sleep changes, REM sleep was inhibited and animals were febrile. Gram-negative bacteria, such as Escherichia coli or Pasteurella multocida, also induce biphasic NREM sleep responses and REM sleep inhibition, although the timing and magnitude of these effects depend on route of administration (Opp and Toth, 2003).
The first bacterial component demonstrated to alter sleep was a specific muramyl peptide derived from bacterial cell wall peptidoglycans (Krueger et al, 1984). Another bacterial product that is involved in sleep responses to gram-negative bacteria is the LPS component of cell wall endotoxin. LPS and has been intensively studied in animal models and humans volunteers with respect to effects on sleep (Mullington et al, 2000; Krueger, 2008). Healthy human volunteers injected with LPS manifest sleep changes, fever, cytokine expression, and hormonal changes (Mullington and Korth et al, 2000) somewhat similar to those seen in animals. However, the impact of LPS on the human EEG differs from those observed in rabbits or rats, and in humans it requires a higher LPS dose to increase NREM sleep than it does to suppress REM sleep.
Most experimental studies of bacterial infections and sleep have used inoculation of a single pathogenic species as the infectious challenge. The gut microbiome, however, is polymicrobial and many infections result from invasion by multiple pathogen species. Such is the case in sepsis, during which polymicrobial infections are routinely the case. The preclinical sepsis model considered to be the gold standard is cecal ligation and puncture (CLP; (Nemzek et al, 2008)). CLP produces a polymicrobial infection that is considered clinically relevant because of its time course, the dynamic changes in cardiac function, and because there is a progressive release of inflammatory mediators. Sleep is altered during the acute phase of CLP sepsis, which occurs from 1 to 4 days after sepsis induction (Baracchi et al, 2011). During this period, NREM and REM sleep of rats increases during the dark period, whereas these sleep phases are reduced during the light period. Analyses of EEG power spectrums for delta and theta-beta power ratios during wake vs sleep cycles further show that LPS-exposed mice evidence alterations in theta-beta power ratios that are circadian cycle and activity state dependent (Adler et al, 2014).
These changes in sleep coincide with increased cytokine mRNA and protein in brain (Granger et al, 2013). Of interest, effects of sepsis on body temperature and activity rhythms persist long after the animal has recovered and is no longer at risk of dying (Granger et al, 2013). These observations suggest that sepsis alters brain function, and are in agreement with observations that patients surviving sepsis often suffer severe and debilitating cognitive impairment. Aging and sleep status also impact BBB transport of TNF. BBB transport of TNF increases during sepsis in young mice, but not in aged (Opp and Krueger, 2015). Interactions among age, sleep status, and BBB function have received little attention, but are likely to be important determinants of outcomes in old and oldest old persons in response to inflammatory insult.
Cytokines have an independent role in the regulation of sleep, and substantial evidence demonstrates that IL-1 and TNF are involved in physiological sleep regulation and in the changes in sleep that occur during inflammatory diseases and pathologies (Obal and Krueger, 2003; Krueger, 2008). The IL1-receptor antagonist and TNF soluble receptor are normal gene products found in blood and brain, and their concentrations are altered by sleep (Krueger, 2008). IL-1 and TNF mRNA have diurnal rhythms in brain with the highest values being associated with periods of maximum sleep. TNF protein also has a sleep-associated diurnal rhythm in several brain areas and IL-1in cerebrospinal fluid varies with the sleep–wake cycle (Lue et al, 1988).
Administration of either IL-1 or TNF promotes NREM sleep (Obal and Krueger, 2003; Opp, 2005; Krueger, 2008). The increase in NREM sleep after either IL-1or TNF administration is physiological in that sleep remains episodic and is readily reversible. The effects of IL-1 on sleep depend upon dose and the time of day it is given (Opp et al, 1991), and knockout strains of mice that lack either the type I IL-1 receptor (Fang et al, 1998), the 55 kD TNF receptor (Fang et al, 1997), or both of these receptors (Baracchi and Opp, 2008) sleep less than control strains.
NREM sleep increases after sleep deprivation, excessive food intake or acute mild increases in ambient temperature. The somnogenic actions of each of these manipulations are associated with enhanced production of either IL-1 or TNF. After sleep deprivation, circulating IL-1 increases, brain levels of IL-1 mRNA increase, and the NREM sleep rebound that would normally occur after sleep deprivation is greatly attenuated if either IL-1 or TNF is blocked using antibodies or soluble receptors.
IL-1 and TNF act within a complex biochemical network (reviewed by Krueger (2008)). For example, IL-1 and TNF stimulate NF-κB, a DNA-binding protein involved in transcription of hundreds of gene products. Other sleep-altering cytokines, such as acidic fibroblast growth factor, epidermal growth factor, and nerve growth factor also stimulate NF-κB production. NF-κB promotes IL-1 and TNF production and thus forms a positive feedback loop. Sleep deprivation is associated with the activation of NF-κB in the cerebral cortex, basal forebrain cholinergic neurons, and the lateral hypothalamus. Activation of NF-κB also promotes IL-2, IL-6, IL-8, IL-15, and IL-18 production, each of which promotes sleep in rats (Obal and Krueger, 2003; Krueger, 2008) (Figure 10).

Interleukin 1 and serotonin interact at multiple sites in the brain to regulate NREM sleep. A schematic representation of interactions in the brain among interleukin 1 (IL-1), serotonin (also known as 5-hydroxytryptamine (5-HT)), and γ-aminobutyric acid (GABA) that are relevant for the regulation of NREM sleep. In the dorsal raphe nuclei (DRN), where IL-1 microinjections promote NREM sleep, IL-1 reduces the firing rate of wake-active serotonergic neurons by enhancing the inhibitory effects of GABA. In the hypothalamic preoptic area/basal forebrain region (POA/BF), IL-1 stimulates 5-HT release from axon terminals. 5-HT, in turn, inhibits cholinergic neurons involved in cortical activation and stimulates the synthesis of IL-1, which inhibits wake-promoting neurons and activates a subset of sleep-promoting neurons in the POA/BF. IL-1 in the POA/BF is under potent inhibitory homeostatic control by corticosteroids released into the blood by the adrenal cortex. Corticosteroid levels depend on the activity of the hypothalamic–pituitary–adrenal axis, which is stimulated by activation of the 5-HT system. ACh, acetylcholine; ACTH, adrenocorticotropic hormone; CRH, corticotropin-releasing hormone; IPSP, inhibitory postsynaptic potential; PVN, paraventricular nucleus of the hypothalamus.
There is a growing literature demonstrating direct effects of IL-1 and TNF on neural substrates implicated in the regulation of sleep (reviewed by Imeri and Opp (2009)). Some of these mechanisms include interactions with classical neurotransmitters such as glutamate, serotonin, acetylcholine, γ-amino butyric acid, histamine, and dopamine (Imeri and Opp, 2009). For example, IL-1 increases serotonergic activity in brain regions implicated in sleep regulation (Gemma et al, 1997), and an intact serotonergic system is required for the full effects of IL-1 on sleep to manifest (Imeri et al, 1997, 1999). IL-1 inhibits discharge rates of serotonergic (Manfridi et al, 2003; Brambilla et al, 2007) and cholinergic (Brambilla et al, 2010) neurons in brainstem. Within the hypothalamus, IL-1 increases c-Fos (Baker et al, 2005) and inhibits wake-active neurons (Alam et al, 2004). TNF promotes sleep if microinjected into the anterior hypothalamus, whereas injection of a soluble TNF receptor into this area reduces sleep (Kubota et al, 2002).
Intrauterine infection or inflammation in preterm neonates is a known risk for adverse neurological outcomes, including cognitive, motor, and behavioral disabilities (Adler et al, 2014). Our previous data suggest that there is acute fetal brain inflammation in a mouse model of intrauterine exposure to LPS. We hypothesized that the in utero inflammation induced by LPS produces long-term electroencephalogram (EEG) biomarkers of neurodegeneration in the exposed mice that could be determined by using continuous quantitative video/EEG/EMG analyses. A single LPS injection at E17 was performed in pregnant CD1 dams. Control dams were injected with same volumes of saline (LPS n=10, Control n=8). At postnatal age of P90-100, 24-h synchronous video/EEG/EMG recordings were done using a tethered recording system and implanted subdural electrodes. Behavioral state scoring was performed blind to treatment group, on each 10 s EEG epoch using synchronous video, EMG and EEG trace signatures to generate individual hypnograms. Automated EEG power spectrums were analyzed for delta and theta-beta power ratios during wake vs sleep cycles. Both control and LPS hypnograms showed an ultradian wake/sleep cycling. Since rodents are nocturnal animals, control mice showed the expected diurnal variation with significantly longer time spent in wake states during the dark cycle phase. In contrast, the LPS-treated mice lost this circadian rhythm. Sleep microstructure also showed significant alteration in the LPS mice specifically during the dark cycle, caused by significantly longer average NREM cycle durations. No significance was found between treatment groups for the delta power data; however, significant activity-dependent changes in theta-beta power ratios seen in controls were absent in the LPS-exposed mice. In conclusion, exposure to in utero inflammation in CD1 mice resulted in significantly altered sleep architecture as adults that were circadian cycle and activity state dependent (Adler, Ammanuel et al. 2014).
Clinical and Translational Evidence for Immune Regulation of Sleep
Because diseases that plague western societies and constitute major public health crises are inflammatory, extensive clinical and translational research has been devoted to targeting cytokines and inflammatory mediators in patient populations. For example, TNF receptor antagonists have been used to treat patients suffering from RA, psoriasis, major depressive disorders, alcohol dependence, insomnia, and sleep apnea, and IL1 antagonists effectively treat a variety of inflammatory bone and joint diseases and heart failure. Clinically available inhibitors of either IL-1 (eg, the IL-1-receptor antagonist, anakinra) or TNF (eg, the TNF soluble receptor, etanercept) alleviate fatigue and excess sleepiness in humans with pathologies such as sleep apnea or RA (Franklin, 1999; Vgontzas et al, 2004; Omdal and Gunnarsson, 2005).
Relatively few studies have used polysomnography to determine the impact of cytokine antagonists on sleep of patients undergoing treatment for inflammatory diseases. A recent study by Miller et al of treatment-resistant major depression (Weinberger et al, 2015) demonstrates that TNF antagonism may be effective in reducing inflammation, improving depressive symptoms, and consolidating sleep. In this study, polysomnography was used to determine sleep parameters at baseline from 36 patients with treatment-resistant major depression. Subjects then were randomized into a treatment arm, using the TNF antagonist infliximab, or placebo. Markers of inflammation included CRP, TNF, and its soluble receptors. Infliximab treatment reduced spontaneous arousals during the night and increased sleep efficiency, changes in sleep that correlated with reduction in the soluble TNF receptor 1. In addition, blockade of TNF has been found to induce a short-term normalization of REM sleep in alcohol-dependent patients with high levels of REM sleep (Irwin et al, 2009); the decrease in REM sleep correlated with pharmacologic neutralization of biologically active TNF. Zamarron et al (2004) also found that TNF blockade improved sleep continuity and increased sleep depth in patients with RA. Finally, TNF antagonism has been found to reduce daytime sleepiness in patients with sleep apnea (Vgontzas et al, 2004), and it possibly improves other depressive symptoms among those with high levels of inflammation (Raison et al, 2013).
Whereas numerous clinical studies targeting inflammatory processes within the context of pathology demonstrate improvement in sleep disturbances associated with disease, translational studies of healthy human volunteers have not yielded results similar to those obtained from preclinical animal studies. Discrepancies between the preclinical and translational studies of healthy adults may be attributed to species difference and other factors. For example, the group of Born used a placebo-controlled crossover design to determine the effects of the IL1 receptor antagonist anakinra on sleep and memory function in healthy male volunteers (Schmidt et al, 2015). In this translational study, anakinra reduced IL1 production by monocytes, indicating it was effective in the periphery. However, in contrast to preclinical animal studies, anakinra did not alter sleep or memory function, although EEG slow-wave activity during SWS actually increased. Other translational studies targeting the immune to sleep link also suggest that the relationships revealed by extensive preclinical research are not fully supported, and more studies using human subjects are necessary to understand how targeting inflammatory systems during pathology reduces the burden of disease.
BEHAVIORAL TREATMENT OF SLEEP DISTURBANCE: IMPACT ON INFLAMMATION
Among the various treatment options for patients with sleep disturbance and insomnia, pharmacologic, psychological, and behavioral approaches (eg, cognitive behavioral therapy for insomnia, CBT-I) and mind-body or relaxation-based therapies (eg, Tai chi and Yoga) have been found to yield significant improvements in sleep outcomes (Irwin et al, 2008, 2014), with evidence that CBT-I is as effective in treating insomnia complaints as pharmacological treatments (Morin et al, 1999). Here we focus on behavioral and mind–body interventions that target insomnia, because of their potential to modulate certain aspects of the immune system, including innate immune response elements. Unfortunately, there is an absence of empirical randomized controlled trial data, which have examined the effects of pharmacologic treatment of insomnia on inflammatory or other immune outcomes.
CBT-I has a robust efficacy profile in the treatment of insomnia (Irwin et al, 2006; Morin et al, 2006). CBT-I is a multicomponent behavioral intervention that provides sleep education, stimulus control (strengthening associations between bed and sleep), and therapy for anxiety-provoking beliefs about sleep, primarily targets sleep behaviors with effects on arousal mechanisms. However, its implementation in community and primary care settings remains limited because CBT-I is intensive and requires administration by highly trained therapists; (Morin, 2015) hence recent attention has focused on self-care interventions, such as mind–body interventions, which can be exported to the community. Furthermore, because it is thought that hyperarousal mechanisms, including activation of the SNS, contribute to the onset and perpetuation of insomnia (Morin et al, 2003; Nofzinger et al, 2004; Bonnet and Arand, 2010), we and others have hypothesized that mind–body interventions may also be effective in the treatment of insomnia because these approaches primarily target these arousal mechanisms. Indeed, controlled trial evidence provides support for the use of mind–body interventions for sleep disturbances in adults (Andersen et al, 2013; Britton et al, 2012) with evidence that a movement-based mind-body intervention, Tai chi, can improve sleep quality in older adults (Irwin et al, 2008, 2014). Even mind–body interventions that do not incorporate components of physical activity, such as mindfulness meditation, have been found to improve sleep. For example, we have evaluated the treatment efficacy of a mindful awareness practices (MAPs), on insomnia complaints, and found that MAPs can improve sleep quality in older adults with an ES comparable to CBT-I (Black et al, 2015) (Figure 11). MAPs is an evidence-based program, similar to other mind–body interventions (Goyal et al, 2014), which trains one in the systematic practice of attending to moment-by-moment experiences, thoughts, and emotions from a non-judgmental perspective (Brown and Ryan, 2003).

Estimated PSQI levels (±SE) before and after intervention. Significant (P=0.002) differences were found between groups covarying for pre-intervention PSQI score; PSQI, Pittsburgh Sleep Quality Index.
Randomized controlled trials that test the efficacy of CBT-I or mind–body interventions on insomnia also provide an opportunity to evaluate whether remission of insomnia is causally associated with a reversal of inflammation. In contrast, pharmacological treatment that improves insomnia might have confounding influences on measures of inflammation. Indeed, mind–body interventions that target stress-response pathways also influence inflammatory dynamics. Our meta-analyses found that a variety of different approaches including Tai chi, Yoga, and meditation, reduce circulating levels of CRP, for example (Morgan et al, 2014). Building on this work, genomics-based methods have been recently used to assess inflammation. Seven different randomized controls of mind–body therapies (ie, Tai chi, meditation, Yoga) have all been shown to reverse the pattern of leukocyte transcriptional alterations including activation of genes regulated by the proinflammatory NF-κB/Rel family (Bower and Irwin, 2016).
In insomnia patients, CBT-I and the mind–body intervention, Tai chi, can effectively reverse that activation of inflammatory pathways associated with insomnia (Irwin et al, 2014, 2015). In a randomized, controlled, comparative efficacy trial over 4 months with follow-up at 7 and 16 months in 123 older adults with insomnia, we found that CBT-I and TCC were associated with improvements in sleep quality, fatigue, and depressive symptoms as compared with an active control, sleep seminar (SS). In addition, remission of insomnia was associated with reduced proportion of having high CRP (>3.0 pg/ml) at month 16. SS controlled for nonspecific factors (eg, expectation, group, and attention). Indeed, the levels of this marker of inflammatory risk were 50% lower one year after treatment among those who showed insomnia remission (Irwin et al, 2014), which are comparable to the benefits reported with vigorous physical activity (Ford, 2002) or weight loss (Esposito et al, 2003). Similarly, in men on hemodialysis who had sleep problems findings, exercise training on sleep quality and inflammation (Afshar et al, 2011). Notably, decreases in CRP associated with CBT-I and TCC treatment of insomnia were not related to changes in physical activity.
Further research examined whether treatment of insomnia might reverse increase in TLR-4 induced activation of monocyte production of IL-6 and TNF and increase in proinflammatory gene expression programs and the specific pattern of bioinformatically inferred TF activation (ie, increased activity of NF-κB/Rel and AP-1 TFs) relative to SS. TCC targeted stress effector mechanisms and reduced TLR-4-activated monocyte production of IL-6 and TNF (Figure 12). Along with remission of insomnia, CBT-I was found to reverse the activation of molecular inflammatory signaling pathways, including reduced activity of NF-κB, reduced activity of AP-1, and reduced activity of CREB, due to downregulation of genes primarily from monocytes and dendritic cell populations (Figure 13). Interestingly, the reduction of cellular inflammation induced by TCC was maintained during follow-up at 7 and 16 months, even though improvements in sleep disturbance were not maintained in the long term, suggesting that these changes are independent of improvements in sleep but rather through changes in other effector mechanisms such as the sympovagal balance.

Toll-like 4 Receptor Stimulated Monocytic Production from Baseline to Month 16, by Treatment Group. Values are mean (SEM) percentage of monocytes producing IL-6 (a), TNF (b); or both IL-6 and TNF (c). Shaded area indicates period of administration of intervention following baseline assessment. Significant pairwise comparisons: CBT vs SS P<0.05; TCC vs SS P<0.05, CBT vs TCC P<0.05.

Transcription factor activity as measured by TELiS promoter-based bioinformatic analyses of genes at 4 months (post intervention), showing differential change in gene expression for comparisons of CBT vs SS, and TCC vs SS.
Finally, the focus on inflammation should be viewed within the broader context of sleep disturbances being linked to increased morbidity and mortality, and other biomarkers of diabetes and cardiovascular disease risk. Indeed, in a population-based sample of 109 older adults with chronic and primary insomnia, multisystem biological risk was significantly reduced along with treatment of insomnia (Carroll et al, 2015). Multisystem biological risk was comprised of eight biomarkers: high-density lipoprotein, low-density lipoprotein, triglycerides, hemoglobin A1c, glucose, insulin, CRP, and fibrinogen, in which a multisystem risk score was computed using clinical laboratory cutoffs defined as abnormal. Again, participants assigned to either CBT-I or TCC showed significantly lower risk scores as compared with SS at 16 months. CBT-I reduced risk of being in the high-risk group at 4 months and at 16 months, whereas TCC reduced the risk at 16-months. Improvements in sleep quality, as defined by a clinical severity threshold, reduced the likelihood of being in the high-risk group at 16-months. (Carroll and Seeman et al, 2015).
Given that these clinical biomarkers are associated with cardiovascular, metabolic, and inflammatory disease risk, improving sleep quality has the potential to reduce the risk of chronic disease in older adults with insomnia. Moreover in relation to inflammatory dynamics, behavioral approaches might provide a strategy for redirecting the leukocyte transcriptome via the induction of multiple trans-acting transcription factors via β-adrenergic receptor signaling that is activated in association with life adversity and sleep loss. Together, these data provide an evidence-based molecular framework to understand how behavioral interventions that target sleep may reduce inflammation. Finally, treatment of sleep disturbance may represent a third pillar, along with diet and physical activity, to promote lifestyle changes with benefits on health.
FUTURE DIRECTIONS AND CLINICAL IMPLICATIONS
Investigation of the environmental and personalized genomic inputs that influence sleep and inflammatory biological mechanisms is needed to understand how distinct aspects of sleep map onto immunological signatures. Sleep is not homogeneous, and multiple factors including circadian rhythms, homeostatic drive to sleep, and physical activity, can all impact sleep quality, sleep duration, and sleep depth, yet the vast majority of human research linking sleep disturbance to inflammation has not considered the overlapping influences of these additional factors on sleep processes. In addition, most naturalistic studies have assessed sleep disturbance using self-report measures, and there is a relative absence of research using quantitative polysomnography to better characterize microstructural changes in sleep that might be associated with sleep quality and inflammatory outcomes. A better knowledge of the objective aspects of sleep disturbance on inflammatory disease risk will inform what specific aspects of sleep might be targeted to moderate the associations between sleep disturbance, inflammation, and adverse health outcomes including depression. Moreover, precise risk profiles that are informed by inflammatory mechanisms will guide development of interventions that target sleep disturbance as one strategy to augment therapeutic control of inflammatory- and neuropsychiatric diseases such as depression.
FUNDING AND DISCLOSURE
The authors declare no conflict of interest.
References
Abedelmalek S, Chtourou H, Aloui A, Aouichaoui C, Souissi N, Tabka Z (2013). Effect of time of day and partial sleep deprivation on plasma concentrations of il-6 during a short-term maximal performance. Eur J Appl Physiol 113: 241–248.
Adler DA, Ammanuel S, Lei J, Dada T, Borbiev T, Johnston MV et al (2014). Circadian cycle-dependent EEG biomarkers of pathogenicity in adult mice following prenatal exposure to in utero inflammation. Neuroscience 275: 305–313.
Afshar R, Emany A, Saremi A, Shavandi N, Sanavi S (2011). Effects of intradialytic aerobic training on sleep quality in hemodialysis patients. Iran J Kidney Dis 5: 119–123.
Alam MN, McGinty D, Bashir T, Kumar S, Imeri L, Opp MR et al (2004). Interleukin-1beta modulates state-dependent discharge activity of preoptic area and basal forebrain neurons: Role in sleep regulation. Eur J Neurosci 20: 207–216.
American Psychiatric Association DSM-5 Task Force (2013) Diagnostic and Statistical Manual of Mental Disorders: DSM-5. American Psychiatric Association: Washington, D.C..
Ancoli-Israel S, Cole R, Alessi C, Chambers M, Moorcroft W, Pollak CP (2003). The role of actigraphy in the study of sleep and circadian rhythms. Sleep 26: 342–392.
Andersen SR, Wurtzen H, Steding-Jessen M, Christensen J, Andersen KK, Flyger H et al (2013). Effect of mindfulness-based stress reduction on sleep quality: results of a randomized trial among danish breast cancer patients. Acta Oncol 52: 336–344.
Andreakos E, Foxwell B, Feldmann M (2004). Is targeting toll-like receptors and their signaling pathway a useful therapeutic approach to modulating cytokine-driven inflammation? Immunol Rev 202: 250–265.
Antoni MH, Lutgendorf SK, Blomberg B, Carver CS, Lechner S, Diaz A et al (2012). Cognitive-behavioral stress management reverses anxiety-related leukocyte transcriptional dynamics. Biol Psychiatry 71: 366–372.
Avitsur R, Padgett DA, Dhabhar FS, Stark JL, Kramer KA, Engler H et al (2003). Expression of glucocorticoid resistance following social stress requires a second signal. J Leukoc Biol 74: 507–513.
Baglioni C, Battagliese G, Feige B, Spiegelhalder K, Nissen C, Voderholzer U et al (2011). Insomnia as a predictor of depression: a meta-analytic evaluation of longitudinal epidemiological studies. J Affect Dis 135: 10–19.
Baker FC, Shah S, Stewart D, Angara C, Gong H, Szymusiak R et al (2005). Interleukin 1beta enhances non-rapid eye movement sleep and increases c-fos protein expression in the median preoptic nucleus of the hypothalamus. Am J Physiol Regul Integr Comp Physiol 288: R998–R1005.
Banks WA (2015). The blood-brain barrier in neuroimmunology: tales of separation and assimilation. Brain Behav Immun 44: 1–8.
Banks WA, Ortiz L, Plotkin SR, Kastin AJ (1991). Human interleukin (il) 1 alpha, murine il-1 alpha and murine il-1 beta are transported from blood to brain in the mouse by a shared saturable mechanism. J Pharmacol Exp Ther 259: 988–996.
Baracchi F, Ingiosi AM, Raymond RM Jr, Opp MR (2011). Sepsis-induced alterations in sleep of rats. Am J Physiol Regul Integr Comp Physiol 301: R1467–R1478.
Baracchi F, Opp MR (2008). Sleep-wake behavior and responses to sleep deprivation of mice lacking both interleukin-1 beta receptor 1 and tumor necrosis factor-alpha receptor 1. Brain Behav Immun 22: 982–993.
Berkenbosch F, deGoij DEC, DelRey A, Besedovsky HO (1989). Neuroendocrine, sympathetic and metabolic responses induced by interleukin-1. Neuroendocrinol 50: 570–576.
Besedovsky L, Lange T, Born J (2012). Sleep and immune function. Pflugers Arch 463: 121–137.
Bierwolf C, Struve K, Marshall L, Born J, Fehm HL (1997). Slow wave sleep drives inhibition of pituitary-adrenal secretion in humans. J Neuroendocrinol 9: 479–484.
Bjurström MF, Olmstead R, Irwin MR (2016). Reciprocal relationship between sleep macrostructure and evening- and morning cellular inflammation in rheumatoid arthritis. Psychosom Med (e-pub ahead of print).
Black DS, O'Reilly GA, Olmstead R, Breen EC, Irwin MR (2015). Mindfulness meditation and improvement in sleep quality and daytime impairment among older adults with sleep disturbances: a randomized clinical trial. JAMA Intern Med 175: 494–501.
Blackburn EH (2000). Telomere states and cell fates. Nature 408: 53–56.
Bluthe RM, Michaud B, Kelley KW, Dantzer R (1996). Vagotomy attenuates behavioural effects of interleukin-1 injected peripherally but not centrally. Neuroreport 7: 1485–1488.
Bluthe RM, Walter V, Parnet P, Laye S, Lestage J, Verrier D et al (1994). Lipopolysaccharide induces sickness behaviour in rats by a vagal mediated mechanism. C R Acad Sci III 317: 499–503.
Bonnet MH, Arand DL (1997). Hyperarousal and insomnia. Sleep Med Rev 1: 97–108.
Bonnet MH, Arand DL (2010). Hyperarousal and insomnia: state of the science. Sleep Med Rev 14: 9–15.
Borbely AA, Achermann P (1999). Sleep homeostasis and models of sleep regulation. J Biol Rhythms 14: 557–568.
Born J, Lange T, Hansen K, Molle M, Fehm HL (1997). Effects of sleep and circadian rhythm on human circulating immune cells. J Immunol 158: 4454–4464.
Boudreau P, Yeh WH, Dumont GA, Boivin DB (2013). Circadian variation of heart rate variability across sleep stages. Sleep 36: 1919–1928.
Bower JE, Irwin MR (2016). Mind-body therapies and control of inflammatory biology: a descriptive review. Brain Behav Immun 51: 1–11.
Brambilla D, Barajon I, Bianchi S, Opp MR, Imeri L (2010). Interleukin-1 inhibits putative cholinergic neurons in vitro and rem sleep when microinjected into the rat laterodorsal tegmental nucleus. Sleep 33: 919–929.
Brambilla D, Franciosi S, Opp MR, Imeri L (2007). Interleukin-1 inhibits firing of serotonergic neurons in the dorsal raphe nucleus and enhances gabaergic inhibitory post-synaptic potentials. Eur J Neurosci 26: 1862–1869.
Breen EC, Rezai AR, Nakajima K, Beall GN, Mitsuyasu RT, Hirano T et al (1990). Infection with hiv is associated with elevated il-6 levels and production. J Immunol 144: 480–484.
Britton WB, Haynes PL, Fridel KW, Bootzin RR (2012). Mindfulness-based cognitive therapy improves polysomnographic and subjective sleep profiles in antidepressant users with sleep complaints. Psychother Psychosom 81: 296–304.
Brown KW, Ryan RM (2003). The benefits of being present: mindfulness and its role in psychological well-being. J Pers Soc Psychol 84: 822–848.
Buysse DJ (2014). Sleep health: can we define it? Does it matter? Sleep 37: 9–17.
Buysse DJ, Reynolds CF 3rd, Monk TH, Berman SR, Kupfer DJ (1989). The pittsburgh sleep quality index: a new instrument for psychiatric practice and research. Psychiatry Res 28: 193–213.
Campisi J, d'Adda di Fagagna F (2007). Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 8: 729–740.
Carroll JE, Carrillo C, Olmstead R, Witarama T, Breen EC, Yokomizo M et al (2015). Sleep deprivation and divergent toll-like receptor-4 activation of cellular inflammation in aging. Sleep 38: 205–211.
Carroll JE, Cole SW, Seeman TE, Breen EC, Witarama T, Arevalo JM et al (2016). Partial sleep deprivation activates the DNA damage response (ddr) and the senescence-associated secretory phenotype (sasp) in aged adult humans. Brain Behav Immun 51: 223–229.
Carroll JE, Esquivel S, Goldberg A, Seeman TE, Effros RB, Dock J et al (2016). Insomnia and telomere length in older adults. Sleep 39: 559–564.
Carroll JE, Irwin MR, Stein Merkin S, Seeman TE (2015). Sleep and multisystem biological risk: a population-based study. PLoS One 10: e0118467.
Carroll JE, Seeman TE, Olmstead R, Melendez G, Sadakane R, Bootzin R et al (2015). Improved sleep quality in older adults with insomnia reduces biomarkers of disease risk: pilot results from a randomized controlled comparative efficacy trial. Psychoneuroendocrinol 55: 184–192.
Castanon-Cervantes O, Wu M, Ehlen JC, Paul K, Gamble KL, Johnson RL et al (2010). Dysregulation of inflammatory responses by chronic circadian disruption. J Immunol 185: 5796–5805.
Chennaoui M, Sauvet F, Drogou C, Van Beers P, Langrume C, Guillard M et al (2011). Effect of one night of sleep loss on changes in tumor necrosis factor alpha (tnf-α) levels in healthy men. Cytokine 56: 318–324.
Cho HJ, Eisenberger NI, Olmstead R, Breen EC, Irwin MR (2016). Preexisting mild sleep disturbance as a vulnerability factor for inflammation-induced depressed mood: a human experimental study. Transl Psychiatry 6: e750.
Cho HJ, Lavretsky H, Olmstead R, Levin MJ, Oxman MN, Irwin MR (2008). Sleep disturbance and depression recurrence in community-dwelling older adults: a prospective study. Am J Psychiatry 165: 1543–1550.
Cho HJ, Seeman TE, Kiefe CI, Lauderdale DS, Irwin MR (2015). Sleep disturbance and longitudinal risk of inflammation: moderating influences of social integration and social isolation in the coronary artery risk development in young adults (cardia) study. Brain Behav Immun 46: 319–326.
Christian LM, Glaser R, Porter K, Malarkey WB, Beversdorf D, Kiecolt-Glaser JK (2011). Poorer self-rated health is associated with elevated inflammatory markers among older adults. Psychoneuroendocrin 36: 1495–1504.
Cole JC, Motivala SJ, Buysse DJ, Oxman MN, Levin MJ, Irwin MR (2006). Validation of a 3-factor scoring model for the pittsburgh sleep quality index in older adults. Sleep 29: 112–116.
Cole SW (2010). Elevating the perspective on human stress genomics. Psychoneuroendocrin 35: 955–962.
Cole SW, Korin YD, Fahey JL, Zack JA (1998). Norepinephrine accelerates hiv replication via protein kinase a-dependent effects on cytokine production. J Immunol 161: 610–616.
Collado-Hidalgo A, Sung C, Cole S (2006). Adrenergic inhibition of innate anti-viral response: Pka blockade of type i interferon gene transcription mediates catecholamine support for hiv-1 replication. Brain Behav Immun 20: 552–563.
Coppe JP, Desprez PY, Krtolica A, Campisi J (2010a). The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 5: 99–118.
Coppe JP, Patil CK, Rodier F, Krtolica A, Beausejour CM, Parrinello S et al (2010b). A human-like senescence-associated secretory phenotype is conserved in mouse cells dependent on physiological oxygen. PLoS One 5: e9188.
Cribbet MR, Carlisle M, Cawthon RM, Uchino BN, Williams PG, Smith TW et al (2014). Cellular aging and restorative processes: subjective sleep quality and duration moderate the association between age and telomere length in a sample of middle-aged and older adults. Sleep 37: 65–70.
Dantzer R (2012). Depression and inflammation: an intricate relationship. Biol Psychiatry 71: 4–5.
Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW (2008). From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 9: 46–56.
de Kloet ER, Oitzl MS, Schobitz B (1994). Cytokines and the grain corticosteroid receptor balance: relevance to pathopysiology of neuroendocrine-immune communication. Psychoneuroendocrin 19: 121–134.
De Zambotti M, Covassin N, De Min Tona G, Sarlo M, Stegagno L (2011). Sleep onset and cardiovascular activity in primary insomnia. J Sleep Res 20: 318–325.
Dew MA, Hoch CC, Buysse DJ, Monk TH, Begely AE, Houck PR et al (2003). Healthy older adults' sleep predicts all-cause mortality at 4 to 19 years of follow-up. Psychosom Med 65: 63–73.
Dew MA, Reynolds CF 3rd, Buysse DJ, Houck PR, Hoch CC, Monk TH et al (1996). Electroencephalographic sleep profiles during depression. effects of episode duration and other clinical and psychosocial factors in older adults. Arch Gen Psychiatry 53: 148–156.
Dimitrov S, Besedovsky L, Born J, Lange T (2015). Differential acute effects of sleep on spontaneous and stimulated production of tumor necrosis factor in men. Brain Behav Immun 47: 201–210.
Dimitrov S, Lange T, Benedict C, Nowell MA, Jones SA, Scheller J et al (2006). Sleep enhances il-6 trans-signaling in humans. FASEB J 20: 2174–2176.
Dinarello CA (2009). Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol 27: 519–550.
Drake CL, Roehrs TA, Royer H, Koshorek G, Turner RB, Roth T (2000). Effects of an experimentally induced rhinovirus cold on sleep, performance, and daytime alertness. Physiol Behav 71: 75–81.
Dryman A, Eaton WW (1991). Affective symptoms associated with the onset of major depression in the community: findings from the us national institute of mental health epidemiologic catchment area program. Acta Psychiatr Scand 84: 1–5.
Eisenberger NI, Berkman ET, Inagaki TK, Rameson LT, Mashal NM, Irwin MR (2010a). Inflammation-induced anhedonia: endotoxin reduces ventral striatum responses to reward. Biol Psychiatry 68: 748–754.
Eisenberger NI, Inagaki TK, Mashal NM, Irwin MR (2010a). Inflammation and social experience: an inflammatory challenge induces feelings of social disconnection in addition to depressed mood. Brain Behav Immun 24: 558–563.
Eisenberger NI, Inagaki TK, Rameson LT, Mashal NM, Irwin MR (2009). An fmri study of cytokine-induced depressed mood and social pain: the role of sex differences. Neuroimage 47: 881–890.
Esposito K, Pontillo A, Di Palo C, Giugliano G, Masella M, Marfella R et al (2003). Effect of weight loss and lifestyle changes on vascular inflammatory markers in obese women: a randomized trial. JAMA 289: 1799–1804.
Fang J, Sanborn CK, Renegar KB, Majde JA, Krueger JM (1995). Influenza viral infections enhance sleep in mice. Proc Soc Exper Biol Med 210: 242–252.
Fang J, Wang Y, Krueger JM (1997). Mice lacking the tnf 55 kda receptor fail to sleep more after tnfalpha treatment. J Neurosci 17: 5949–5955.
Fang J, Wang Y, Krueger JM (1998). Effects of interleukin-1 beta on sleep are mediated by the type i receptor. Am J Physiol 274: R655–R660.
Faraut B, Boudjeltia KZ, Dyzma M, Rousseau A, David E, Stenuit P et al (2011). Benefits of napping and an extended duration of recovery sleep on alertness and immune cells after acute sleep restriction. Brain Behav Immun 25: 16–24.
Festa A, D'Agostino R Jr, Howard G, Mykkanen L, Tracy RP, Haffner SM (2000). Chronic subclinical inflammation as part of the insulin resistance syndrome: The insulin resistance atherosclerosis study (iras). Circulation 102: 42–47.
Ford DE, Kamerow DB (1989). Epidemiologic study of sleep disturbances and psychiatric disorders. An opportunity for prevention? JAMA 262: 1479–1484.
Ford ES (2002). Does exercise reduce inflammation? Physical activity and c-reactive protein among U.S. Adults. Epidemiology 13: 561–568.
Franceschi C, Campisi J (2014). Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci 69 (Suppl 1): S4–S9.
Franklin CM (1999). Clinical experience with soluble tnf p75 receptor in rheumatoid arthritis. Semin Arthritis Rheum 29: 172–181.
Franzen PL, Buysse DJ, Rabinovitz M, Pollock BG, Lotrich FE (2010). Poor sleep quality predicts onset of either major depression or subsyndromal depression with irritability during interferon-alpha treatment. Psychiatry Res 177: 240–245.
Fredrickson BL, Grewen KM, Coffey KA, Algoe SB, Firestine AM, Arevalo JM et al (2013). A functional genomic perspective on human well-being. Proc Natl Acad Sci USA 110: 13684–13689.
Freund A, Orjalo AV, Desprez PY, Campisi J (2010). Inflammatory networks during cellular senescence: causes and consequences. Trends Mol Med 16: 238–246.
Frey DJ, Fleshner M, Wright KP Jr (2007). The effects of 40 h of total sleep deprivation on inflammatory markers in healthy young adults. Brain Behav Immun 21: 1050–1057.
Friedman EM, Hayney MS, Love GD, Urry HL, Rosenkranz MA, Davidson RJ et al (2005). Social relationships, sleep quality, and interleukin-6 in aging women. Proc Natl Acad Sci U S A 102: 18757–18762.
Gao B, Wang H, Lafdil F, Feng D (2012). Stat proteins - key regulators of anti-viral responses, inflammation, and tumorigenesis in the liver. J Hepatol 57: 430–441.
Gemma C, Imeri L, de Simoni MG, Mancia M (1997). Interleukin-1 induces changes in sleep, brain temperature, and serotonergic metabolism. Am J Physiol 272: R601–R606.
Gimeno D, Kivimaki M, Brunner EJ, Elovainio M, De Vogli R, Steptoe A et al (2009). Associations of c-reactive protein and interleukin-6 with cognitive symptoms of depression: 12-year follow-up of the whitehall ii study. Psychol Med 39: 413–423.
Gourmelon P, Briet D, Clarencon D, Court L, Tsiang H (1991). Sleep alterations in experimental street rabies virus infection occur in the absence of major EEG abnormalities. Brain Res 554: 159–165.
Goyal M, Singh S, Sibinga EM, Gould NF, Rowland-Seymour A, Sharma R et al (2014). Meditation programs for psychological stress and well-being: a systematic review and meta-analysis. JAMA Intern Med 174: 357–368.
Grandner MA, Buxton OM, Jackson N, Sands-Lincoln M, Pandey A, Jean-Louis G (2013). Extreme sleep durations and increased c-reactive protein: effects of sex and ethnoracial group. Sleep 36: 769–779E.
Granger JI, Ratti PL, Datta SC, Raymond RM, Opp MR (2013). Sepsis-induced morbidity in mice: effects on body temperature, body weight, cage activity, social behavior and cytokines in brain. Psychoneuroendocrinology 38: 1047–1057.
Gutierrez EG, Banks WA, Kastin AJ (1993). Murine tumor necrosis factor alpha is transported from blood to brain in the mouse. J Neuroimmunol 47: 169–176.
Haack M, Sanchez E, Mullington JM (2007). Elevated inflammatory markers in response to prolonged sleep restriction are associated with increased pain experience in healthy volunteers. Sleep 30: 1145–1152.
Hansen MK, Krueger JM (1997). Subdiaphragmatic vagotomy blocks the sleep- and fever-promoting effects of interleukin-1beta. Am J Physiol 273: R1246–R1253.
Hayashino Y, Jackson JL, Hirata T, Fukumori N, Nakamura F, Fukuhara S et al (2014). Effects of exercise on c-reactive protein, inflammatory cytokine and adipokine in patients with type 2 diabetes: a meta-analysis of randomized controlled trials. Metabolism 63: 431–440.
He J, Hsuchou H, He Y, Kastin AJ, Wang Y, Pan W (2014). Sleep restriction impairs blood-brain barrier function. J Neurosci 34: 14697–14706.
Herrero L, Valcarcel L, da Silva CA, Albert N, Diez-Noguera A, Cambras T et al (2015). Altered circadian rhythm and metabolic gene profile in rats subjected to advanced light phase shifts. PLoS One 10: e0122570.
Imeri L, Bianchi S, Mancia M (1997). Muramyl dipeptide and il-1 effects on sleep and brain temperature after inhibition of serotonin synthesis. Am J Physiol 273: R1663–R1668.
Imeri L, Mancia M, Opp MR (1999). Blockade of 5-hydroxytryptamine (serotonin)-2 receptors alters interleukin-1-induced changes in rat sleep. Neuroscience 92: 745–749.
Imeri L, Opp MR (2009). How (and why) the immune system makes us sleep. Nat Rev Neurosci 10: 199–210.
Irwin M, Gillin JC, Dang J, Weissman J, Phillips E, Ehlers CL (2002). Sleep deprivation as a probe of homeostatic sleep regulation in primary alcoholics. Biol Psychiatry 51: 632–641.
Irwin M, Olmstead R, Breen E, Witarama T, Carrillo C, Sadeghi N et al (2014). Tai chi, cellular inflammation, and transcriptome dynamics in breast cancer survivors with insomnia: a randomized controlled trial. J Natl Cancer Inst 50: 295–301.
Irwin M, Rinetti G, Redwine L, Motivala S, Dang J, Ehlers C (2004). Nocturnal proinflammatory cytokine-associated sleep disturbances in abstinent African American alcoholics. Brain Behav Immun 18: 349–360.
Irwin M, Thompson J, Miller C, Gillin JC, Ziegler M (1999). Effects of sleep and sleep deprivation on catecholamine and interleukin-2 levels in humans: clinical implications. J Clin Endocrin Metab 84: 1979–1985.
Irwin MR (2015). Why sleep is important for health: a psychoneuroimmunology perspective. Ann Rev Psychol 66: 2.1–2.30.
Irwin MR, Carrillo C, Olmstead R (2010). Sleep loss activates cellular markers of inflammation: sex differences. Brain Behav Immun 24: 54–57.
Irwin MR, Cole JC, Nicassio PM (2006). Comparative meta-analysis of behavioral interventions for insomnia and their efficacy in middle-aged adults and in older adults 55+ years of age. Health Psychol 25: 3–14.
Irwin MR, Cole SW (2011). Reciprocal regulation of the neural and innate immune systems. Nature Rev Immunol 11: 625–632.
Irwin MR, Olmstead R, Breen EC, Witarama T, Carrillo C, Sadeghi N et al (2015). Cognitive behavioral therapy and Tai Chi reverse cellular and genomic markers of inflammation in late-life insomnia: a randomized controlled trial. Biol Psychiatry 78: 721–729.
Irwin MR, Olmstead R, Carrillo C, Sadeghi N, Breen EC, Witarama T et al (2014). Cognitive behavioral therapy vs Tai chi for late life insomnia and inflammatory risk: a randomized controlled comparative efficacy trial. Sleep 37: 1543–1552.
Irwin MR, Olmstead R, Carrillo C, Sadeghi N, Fitzgerald JD, Ranganath VK et al (2012). Sleep loss exacerbates fatigue, depression, and pain in rheumatoid arthritis. Sleep 35: 537–543.
Irwin MR, Olmstead R, Carroll JE (2016). Sleep disturbance, sleep duration, and inflammation: a systematic review and meta-analysis of cohort studies and experimental sleep deprivation. Biol Psychiatry 80: 40–52.
Irwin MR, Olmstead R, Motivala SJ (2008). Improving sleep quality in older adults with moderate sleep complaints: a randomized controlled trial of Tai Chi chih. Sleep 31: 1001–1008.
Irwin MR, Olmstead R, Valladares EM, Breen EC, Ehlers CL (2009). Tumor necrosis factor antagonism normalizes rapid eye movement sleep in alcohol dependence. Biol Psychiatry 66: 191–195.
Irwin MR, Wang M, Campomayor CO, Collado-Hidalgo A, Cole S (2006). Sleep deprivation and activation of morning levels of cellular and genomic markers of inflammation. Arch Intern Med 166: 1756–1762.
Irwin MR, Wang M, Ribeiro D, Cho HJ, Olmstead R, Breen EC et al (2008). Sleep loss activates cellular inflammatory signaling. Biol Psychiatry 64: 538–540.
Irwin MR, Witarama T, Caudill M, Olmstead R, Breen EC (2015). Sleep loss activates cellular inflammation and signal transducer and activator of transcription (stat) family proteins in humans. Brain Behav Immun 47: 86–92.
Irwin MR, Ziegler M (2005). Sleep deprivation potentiates activation of cardiovascular and catecholamine responses in abstinent alcoholics. Hypertension 45: 252–257.
Ishihara K, Hirano T (2002). Il-6 in autoimmune disease and chronic inflammatory proliferative disease. Cytokine Growth Factor Rev 13: 357–368.
Jarcho MR, Slavich GM, Tylova-Stein H, Wolkowitz OM, Burke HM (2013). Dysregulated diurnal cortisol pattern is associated with glucocorticoid resistance in women with major depressive disorder. Biol Psychol 93: 150–158.
Jaussent I, Bouyer J, Ancelin ML, Akbaraly T, Peres K, Ritchie K et al (2011). Insomnia and daytime sleepiness are risk factors for depressive symptoms in the elderly. Sleep 34: 1103–1110.
Kapas L, Bohnet SG, Traynor TR, Majde JA, Szentirmai E, Magrath P et al (2008). Spontaneous and influenza virus-induced sleep are altered in tnf-alpha double-receptor deficient mice. J Appl Physiol (1985) 105: 1187–1198.
Karin M (2006). Nuclear factor-kappab in cancer development and progression. Nature 441: 431–436.
Kattler H, Dijk DJ, Borbely AA (1994). Effect of unilateral somatosensory stimulation prior to sleep on the sleep EEG in humans. J Sleep Res 3: 159–164.
Kennedy BK, Berger SL, Brunet A, Campisi J, Cuervo AM, Epel ES et al (2014). Geroscience: linking aging to chronic disease. Cell 159: 709–713.
Kierlin L, Olmstead R, Yokomizo M, Nicassio P, Irwin MR (2012). Diagnostic and statistical manual criteria for insomnia related impairment in daytime functioning: polysomnographic correlates in older adults. Sleep Med 13: 958–960.
Krueger JM (2008). The role of cytokines in sleep regulation. Curr Pharm Des 14: 3408–3416.
Krueger JM, Karnovsky ML, Martin SA, Pappenheimer JR, Walter J, Biemann K (1984). Peptidoglycans as promoters of slow-wave sleep. Ii. Somnogenic and pyrogenic activities of some naturally occurring muramyl peptides; correlations with mass spectrometric structure determination. J Biol Chem 259: 12659–12662.
Krueger JM, Obal F (1993). Growth hormone releasing hormone and interleukin-1 in sleep regulation. FASEB J 7: 645–652.
Krueger JM, Toth LA, Floyd R, Fang J, Kapas L, Bredow S et al (1994). Sleep, microbes and cytokines. Neuroimmunomodulation 1: 100–109.
Kubota T, Li N, Guan Z, Brown RA, Krueger JM (2002). Intrapreoptic microinjection of tnf-alpha enhances non-rem sleep in rats. Brain Res 932: 37–44.
Lanfranchi PA, Pennestri MH, Fradette L, Dumont M, Morin CM, Montplaisir J (2009). Nighttime blood pressure in normotensive subjects with chronic insomnia: implications for cardiovascular risk. Sleep 32: 760–766.
Lange T, Dimitrov S, Fehm HL, Born J (2006). Sleep-like concentrations of growth hormone and cortisol modulate type1 and type2 in vitro cytokine production in human t cells. Int Immunopharmacol 6: 216–225.
Larkin EK, Rosen CL, Kirchner HL, Storfer-Isser A, Emancipator JL, Johnson NL et al (2005). Variation of c-reactive protein levels in adolescents: association with sleep-disordered breathing and sleep duration. Circulation 111: 1978–1984.
Laye S, Bluthe RM, Kent S, Combe C, Medina C, Parnet P et al (1995). Subdiaphragmatic vagotomy blocks induction of il-1 beta mrna in mice brain in response to peripheral lps. Am J Physiol 268: R1327–R1331.
Lee E, Cho HJ, Olmstead R, Levin MJ, Oxman MN, Irwin MR (2013). Persistent sleep disturbance: a risk factor for recurrent depression in community-dwelling older adults. Sleep 36: 1685–1691.
Lee JH, Kim SJ, Jung HH (2009). Nocturnal sleep related with metabolic markers in end-stage renal disease patients receiving hemodialysis. Psychiatry Investig 6: 34–38.
Levy DE, Lee CK (2002). What does stat3 do? J Clin Invest 109: 1143–1148.
Littner M, Hirshkowitz M, Kramer M, Kapen S, Anderson WM, Bailey D et al. American Academy of Sleep M,Standards of Practice C (2003). Practice parameters for using polysomnography to evaluate insomnia: an update. Sleep 26: 754–760.
Liu Y, Sanoff HK, Cho H, Burd CE, Torrice C, Ibrahim JG et al (2009). Expression of p16(ink4a) in peripheral blood t-cells is a biomarker of human aging. Aging Cell 8: 439–448.
Liu Y, Wheaton AG, Chapman DP, Croft JB (2013). Sleep duration and chronic diseases among U.S. adults age 45 years and older: evidence from the 2010 behavioral risk factor surveillance system. Sleep 36: 1421–1427.
Liukkonen T, Rasanen P, Ruokonen A, Laitinen J, Jokelainen J, Leinonen M et al (2007). C-reactive protein levels and sleep disturbances: observations based on the Northern Finland 1966 birth cohort study. Psychosom Med 69: 756–761.
Lotrich FE, Germain A (2015). Decreased delta sleep ratio and elevated alpha power predict vulnerability to depression during interferon-alpha treatment. Acta Neuropsychiatr 27: 14–24.
Lue FA, Bail M, Jephthah-Ochola J, Carayanniotis K, Gorczynski R, Moldofsky H (1988). Sleep and cerebrospinal fluid interleukin-1-like activity in the cat. Inter J Neurosci 42: 179–183.
Machida M, Ambrozewicz MA, Breving K, Wellman LL, Yang L, Ciavarra RP et al (2014). Sleep and behavior during vesicular stomatitis virus induced encephalitis in balb/cj and c57bl/6j mice. Brain Behav Immun 35: 125–134.
Manfridi A, Brambilla D, Bianchi S, Mariotti M, Opp MR, Imeri L (2003). Interleukin-1beta enhances non-rapid eye movement sleep when microinjected into the dorsal raphe nucleus and inhibits serotonergic neurons in vitro. Eur J Neurosci 18: 1041–1049.
Marsland AL, Gianaros PJ, Abramowitch SM, Manuck SB, Hariri AR (2008). Interleukin-6 covaries inversely with hippocampal grey matter volume in middle-aged adults. Biol Psychiatry 64: 484–490.
Medzhitov R (2008). Origin and physiological roles of inflammation. Nature 454: 428–435.
Meier-Ewert HK, Ridker PM, Rifai N, Regan MM, Price NJ, Dinges DF et al (2004). Effect of sleep loss on c-reactive protein, an inflammatory marker of cardiovascular risk. J Am Coll Cardiol 43: 678–683.
Miklossy G, Hilliard TS, Turkson J (2013). Therapeutic modulators of stat signalling for human diseases. Nat Rev Drug Discov 12: 611–629.
Miller GE, Cohen S, Ritchey AK (2002). Chronic psychological stress and the regulation of pro-inflammatory cytokines: a glucocorticoid-resistance model. Health Psychol 21: 531–541.
Miller MA, Kandala NB, Kivimaki M, Kumari M, Brunner EJ, Lowe GD et al (2009). Gender differences in the cross-sectional relationships between sleep duration and markers of inflammation: Whitehall II study. Sleep 32: 857–864.
Moieni M, Irwin MR, Jevtic I, Olmstead R, Breen EC, Eisenberger NI (2015). Sex differences in depressive and socioemotional responses to an inflammatory challenge: implications for sex differences in depression. Neuropsychopharmacology 40: 1709–1716.
Moldofsky H, Lue FA, Davidson JR, Gorczynski R (1989). Effects of sleep deprivation on human immune functions. FASEB J 3: 1972–1977.
Morgan N, Irwin MR, Chung M, Wang C (2014). The effects of mind-body therapies on the immune system: meta-analysis. PLoS One 9: e100903.
Morin C, Cholecchi C, Stone J, Sood R, Brink D (1999). Behavioral and pharmacological therapies for late-life insomnia: a randomized controlled trial. JAMA 281: 991–999.
Morin CM (2015). Cognitive behavioral therapy for chronic insomnia: state of the science versus current clinical practices. Ann Intern Med 163: 236–237.
Morin CM, Belleville G, Belanger L, Ivers H (2011). The insomnia severity index: psychometric indicators to detect insomnia cases and evaluate treatment response. Sleep 34: 601–608.
Morin CM, Bootzin RR, Buysse DJ, Edinger JD, Espie CA, Lichstein KL (2006). Psychological and behavioral treatment of insomnia: update of the recent evidence (1998-2004). Sleep 29: 1398–1414.
Morin CM, Hauri PJ, Espie CA, Spielman AJ, Buysse DJ, Bootzin RR (1999). Nonpharmacologic treatment of chronic insomnia. An american academy of sleep medicine review. Sleep 22: 1134–1156.
Morin CM, Rodrigue S, Ivers H (2003). Role of stress, arousal, and coping skills in primary insomnia. Psychosom Med 65: 259–267.
Mullington J, Korth C, Hermann DM, Orth A, Galanos C, Holsboer F et al (2000). Dose-dependent effects of endotoxin on human sleep. Am J Physiol Regul Integr Comp Physiol 278: R947–R955.
Nance DM, Sanders VM (2007). Autonomic innervation and regulation of the immune system (1987-2007). Brain Behav Immun 21: 736–745.
Neale EP, Batterham MJ, Tapsell LC (2016). Consumption of a healthy dietary pattern results in significant reductions in c-reactive protein levels in adults: a meta-analysis. Nutr Res 36: 391–401.
Nemzek JA, Hugunin KM, Opp MR (2008). Modeling sepsis in the laboratory: merging sound science with animal well-being. Comp Med 58: 120–128.
Nofzinger EA, Buysse DJ, Germain A, Price JC, Miewald JM, Kupfer DJ (2004). Functional neuroimaging evidence for hyperarousal in insomnia. Am J Psychiatry 161: 2126–2128.
Obal F Jr, Krueger JM (2003). Biochemical regulation of non-rapid-eye-movement sleep. Front Biosci 8: d520–d550.
Ohayon MM (2002). Epidemiology of insomnia: what we know and what we still need to learn. Sleep Med Rev 6: 97–111.
Ohayon MM (2005). Prevalence and correlates of nonrestorative sleep complaints. Arch Intern Med 165: 35–41.
Ohayon MM, Carskadon MA, Guilleminault C, Vitiello MV (2004). Meta-analysis of quantitative sleep parameters from childhood to old age in healthy individuals: developing normative sleep values across the human lifespan. Sleep 27: 1255–1273.
Ohayon MM, Riemann D, Morin C, Reynolds CF 3rd (2012). Hierarchy of insomnia criteria based on daytime consequences. Sleep Med 13: 52–57.
Olofsson PS, Rosas-Ballina M, Levine YA, Tracey KJ (2012). Rethinking inflammation: neural circuits in the regulation of immunity. Immunol Rev 248: 188–204.
Omdal R, Gunnarsson R (2005). The effect of interleukin-1 blockade on fatigue in rheumatoid arthritis—a pilot study. Rheumatol Int 25: 481–484.
Opp MR (2005). Cytokines and sleep. Sleep Med Rev 9: 355–364.
Opp MR, Imeri L (1999). Sleep as a behavioral model of neuro-immune interactions. Acta Neurobiol Exp (Wars) 59: 45–53.
Opp MR, Krueger JM (2015). Sleep and immunity: a growing field with clinical impact. Brain Behav Immun 47: 1–3.
Opp MR, Obal F Jr, Krueger JM (1991). Interleukin 1 alters rat sleep: temporal and dose-related effects. Am J Physiol 260: R52–R58.
Opp MR, Toth LA (1998). Somnogenic and pyrogenic effects of interleukin-1beta and lipopolysaccharide in intact and vagotomized rats. Life Sci 62: 923–936.
Opp MR, Toth LA (2003). Neural-immune interactions in the regulation of sleep. Front Biosci 8: D768–D779.
Pace TW, Hu F, Miller AH (2007). Cytokine-effects on glucocorticoid receptor function: Relevance to glucocorticoid resistance and the pathophysiology and treatment of major depression. Brain Behav Immun 21: 9–19.
Pan W, Kastin AJ (2016). The blood-brain barrier: Regulatory roles in wakefulness and sleep. Neuroscientist e-pub ahead of print.
Patel SR, Zhu X, Storfer-Isser A, Mehra R, Jenny NS, Tracy R et al (2009). Sleep duration and biomarkers of inflammation. Sleep 32: 200–204.
Plotkin SR, Banks WA, Kastin AJ (1996). Comparison of saturable transport and extracellular pathways in the passage of interleukin-1 alpha across the blood-brain barrier. J Neuroimmunol 67: 41–47.
Prather AA, Epel ES, Cohen BE, Neylan TC, Whooley MA (2013). Gender differences in the prospective associations of self-reported sleep quality with biomarkers of systemic inflammation and coagulation: findings from the heart and soul study. J Psychiatr Res 47: 1228–1235.
Prather AA, Puterman E, Lin J, O'Donovan A, Krauss J, Tomiyama AJ et al (2011). Shorter leukocyte telomere length in midlife women with poor sleep quality. J Aging Res 2011: 721390.
Quan N, Whiteside M, Herkenham M (1998). Time course and localization patterns of interleukin-1beta messenger rna expression in brain and pituitary after peripheral administration of lipopolysaccharide. Neuroscience 83: 281–293.
Raison CL, Miller AH (2013). Role of inflammation in depression: implications for phenomenology, pathophysiology and treatment. Mod Trends Pharmacopsychiatri 28: 33–48.
Raison CL, Rutherford RE, Woolwine BJ, Shuo C, Schettler P, Drake DF et al (2013). A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: the role of baseline inflammatory biomarkers. JAMA Psychiatry 70: 31–41.
Ramey SL, Perkhounkova Y, Moon M, Budde L, Tseng HC, Clark MK (2012). The effect of work shift and sleep duration on various aspects of police officers' health. Workplace Health Saf 60: 215–222.
Rechtschaffen A, Kales A (1968) A Manual of Standardized Terminology, Techniques and Scoring System for Sleep Stages of Human Subjects. Bethesda, N Inst Neurol Dis & Blindness: Bethesda.
Redwine L, Hauger RL, Gillin JC, Irwin M (2000). Effects of sleep and sleep deprivation on interleukin-6, growth hormone, cortisol, and melatonin levels in humans. J Clin Endocrinol Metab 85: 3597–3603.
Reynolds CF, Kupfer DJ, Hoch CC, Houck PR, Stack JA, Beman SR et al (1987). Sleep deprivation as a probe in the elderly. Arch Gen Psychiatry 44: 982–990.
Ridker PM, Buring JE, Cook NR, Rifai N (2003). C-reactive protein, the metabolic syndrome, and risk of incident cardiovascular events: an 8-year follow-up of 14 719 initially healthy american women. Circulation 107: 391–397.
Ridker PM, Cook N (2004). Clinical usefulness of very high and very low levels of c-reactive protein across the full range of framingham risk scores. Circulation 109: 1955–1959.
Rief W, Mills PJ, Ancoli-Israel S, Ziegler MG, Pung MA, Dimsdale JE (2010). Overnight changes of immune parameters and catecholamines are associated with mood and stress. Psychosom Med 72: 755–762.
Riemann D (2010). Hyperarousal and insomnia: state of the science. Sleep Med Rev 14: 17.
Sahin E, Colla S, Liesa M, Moslehi J, Muller FL, Guo M et al (2011). Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 470: 359–365.
Sato S, Sakurai T, Ogasawara J, Takahashi M, Izawa T, Imaizumi K et al (2014). A circadian clock gene, rev-erbalpha, modulates the inflammatory function of macrophages through the negative regulation of ccl2 expression. J Immunol 192: 407–417.
Satoh M, Shimoda Y, Maesawa C, Akatsu T, Ishikawa Y, Minami Y et al (2005). Activated toll-like receptor 4 in monocytes is associated with heart failure after acute myocardial infarction. Int J Cardiol. 109: 226–234.
Sauvet F, Leftheriotis G, Gomez-Merino D, Langrume C, Drogou C, Van Beers P et al (2010). Effect of acute sleep deprivation on vascular function in healthy subjects. J Appl Physiol 108: 68–75.
Schmid SM, Hallschmid M, Jauch-Chara K, Wilms B, Lehnert H, Born J et al (2011). Disturbed glucoregulatory response to food intake after moderate sleep restriction. Sleep 34: 371–377.
Schmidt EM, Linz B, Diekelmann S, Besedovsky L, Lange T, Born J (2015). Effects of an interleukin-1 receptor antagonist on human sleep, sleep-associated memory consolidation, and blood monocytes. Brain Behav Immun 47: 178–185.
Shearer WT, Reuben JM, Mullington JM, Price NJ, Lee BN, Smith EO et al (2001). Soluble TNF-alpha receptor 1 and IL-6 plasma levels in humans subjected to the sleep deprivation model of spaceflight. J Allergy Clin Immunol 107: 165–170.
Shochat T, Umphress J, Israel AG, Ancoli-Israel S (1999). Insomnia in primary care patients. Sleep 22 (Suppl 2): S359–S365.
Shuai K, Liu B (2003). Regulation of jak-stat signalling in the immune system. Nat Rev Immunol 3: 900–911.
Slavich GM, Cole SW (2013). The emerging field of human social genomics. Clin Psychol Sci 1: 331–348.
Slavich GM, Irwin MR (2014). From stress to inflammation and major depressive disorder: a social signal transduction theory of depression. Psychol Bull 140: 774–815.
Smagula SF, Stone KL, Redline S, Ancoli-Israel S, Barrett-Connor E, Lane NE et alOsteoporotic Fractures in Men Research G (2016). Actigraphy- and polysomnography-measured sleep disturbances, inflammation, and mortality among older men. Psychosom Med 78: 686–696.
Smith A (1992) Sleep, colds and performance, in Broughton RJ, Ogilvie R. eds. Birkhauser, Sleep, Arousal, and Performance: A Tribute to Bob Wilkinson. Birkhauser.
Somers VK, Dyken ME, Mark AL, Abboud FM (1993). Sympathetic-nerve activity during sleep in normal subjects. N Engl J Med 328: 303–307.
Spath-Schwalbe E, Uthgenannt D, Voget G, Kern W, Born J, Fehm HL (1993). Corticotropin-releasing hormone-induced adrenocorticotropin and cortisol secretion depends on sleep and wakefulness. J Clin Endocrinol Metab 77: 1170–1173.
Stamatakis KA, Punjabi NM (2010). Effects of sleep fragmentation on glucose metabolism in normal subjects. Chest 137: 95–101.
Stark JL, Avitsur R, Hunzeker J, Padgett DA, Sheridan JF (2002). Interleukin-6 and the development of social disruption-induced glucocorticoid resistance. J Neuroimmunol 124: 9–15.
Stark JL, Avitsur R, Padgett DA, Campbell KA, Beck FM, Sheridan JF (2001). Social stress induces glucocorticoid resistance in macrophages. Am J Physiol Regul Integr Comp Physiol 280: R1799–R1805.
Stenholm S, Kronholm E, Bandinelli S, Guralnik JM, Ferrucci L (2011). Self-reported sleep duration and time in bed as predictors of physical function decline: results from the inchianti study. Sleep 34: 1583–1593.
Steptoe A, Hamer M, Chida Y (2007). The effects of acute psychological stress on circulating inflammatory factors in humans: a review and meta-analysis. Brain Behav Immun 21: 901–912.
Sternberg EM (2006). Neural regulation of innate immunity: a coordinated nonspecific host response to pathogens. Nat Rev Immunol 6: 318–328.
Suarez EC (2008). Self-reported symptoms of sleep disturbance and inflammation, coagulation, insulin resistance and psychosocial distress: evidence for gender disparity. Brain Behav Immun 22: 960–968.
Taheri S, Austin D, Lin L, Nieto FJ, Young T, Mignot E (2007). Correlates of serum c-reactive protein (crp)—no association with sleep duration or sleep disordered breathing. Sleep 30: 991–996.
Tomfohr LM, Edwards KM, Madsen JW, Mills PJ (2015). Social support moderates the relationship between sleep and inflammation in a population at high risk for developing cardiovascular disease. Psychophysiology 52: 1689–1697.
Toth LA (1995). Sleep, sleep deprivation and infectious disease: studies in animals. Adv Neuroimmunol 5: 79–92.
Toth LA, Hughes LF (2004). Macrophage participation in influenza-induced sleep enhancement in c57bl/6j mice. Brain Behav Immun 18: 375–389.
Toth LA, Krueger JM (1988). Alteration of sleep in rabbits by Staphylococcus aureus infection. Infect Immun 56: 1785–1791.
Toth LA, Tolley EA, Broady R, Blakely B, Krueger JM (1994). Sleep during experimental trypanosomiasis in rabbits. Proc Soc Exp Biol Med 205: 174–181.
Toth LA, Verhulst SJ (2003). Strain differences in sleep patterns of healthy and influenza-infected inbred mice. Behav Genet 33: 325–336.
Tracey KJ (2009). Reflex control of immunity. Nat Rev Immunol 9: 418–428.
Trammell RA, Toth LA (2008). Genetic susceptibility and resistance to influenza infection and disease in humans and mice. Expert Rev Mol Diagn 8: 515–529.
Traynor TR, Majde JA, Bohnet SG, Krueger JM (2007). Interferon type i receptor-deficient mice have altered disease symptoms in response to influenza virus. Brain Behav Immun 21: 311–322.
Tubtimtes S, Sukying C, Prueksaritanond S (2009). Sleep problems in out-patient of primary care unit. J Med Assoc Thai 92: 273–278.
Tyring S, Gottlieb A, Papp K, Gordon K, Leonardi C, Wang A et al (2006). Etanercept and clinical outcomes, fatigue, and depression in psoriasis: double-blind placebo-controlled randomised phase III trial. Lancet 367: 29–35.
van Leeuwen WM, Lehto M, Karisola P, Lindholm H, Luukkonen R, Sallinen M et al (2009). Sleep restriction increases the risk of developing cardiovascular diseases by augmenting proinflammatory responses through il-17 and crp. PLoS One 4: e4589.
Vgontzas AN, Fernandez-Mendoza J, Liao D, Bixler EO (2013). Insomnia with objective short sleep duration: the most biologically severe phenotype of the disorder. Sleep Med Rev 17: 241–254.
Vgontzas AN, Liao D, Bixler EO, Chrousos GP, Vela-Bueno A (2009). Insomnia with objective short sleep duration is associated with a high risk for hypertension. Sleep 32: 491–497.
Vgontzas AN, Papanicolaou DA, Bixler EO, Lotsikas A, Zachman K, Kales A et al (1999). Circadian interleukin-6 secretion and quantity and depth of sleep. J Clin Endocrinol Metab 84: 2603–2607.
Vgontzas AN, Pejovic S, Zoumakis E, Lin HM, Bixler EO, Basta M et al (2007). Daytime napping after a night of sleep loss decreases sleepiness, improves performance, and causes beneficial changes in cortisol and interleukin-6 secretion. Am J Physiol Endocrinol Metab 292: E253–E261.
Vgontzas AN, Zoumakis E, Bixler EO, Lin HM, Follett H, Kales A et al (2004). Adverse effects of modest sleep restriction on sleepiness, performance, and inflammatory cytokines. J Clin Endocrinol Metab 89: 2119–2126.
Vgontzas AN, Zoumakis E, Lin HM, Bixler EO, Trakada G, Chrousos GP (2004). Marked decrease in sleepiness in patients with sleep apnea by etanercept, a tumor necrosis factor-alpha antagonist. J Clin Endocrinol Metab 89: 4409–4413.
Vitkovic L, Bockaert J, Jacque C (2000). ‘Inflammatory’ cytokines: neuromodulators in normal brain? J Neurochem 74: 457–471.
Walter JS, Meyers P, Krueger JM (1989). Microinjection of interleukin-1 into brain: separation of sleep and fever responses. Physiol Behav 45: 169–176.
Weinberger JF, Raison CL, Rye DB, Montague AR, Woolwine BJ, Felger JC et al (2015). Inhibition of tumor necrosis factor improves sleep continuity in patients with treatment resistant depression and high inflammation. Brain Behav Immun 47: 193–200.
Wilckens KA, Aizenstein HJ, Nofzinger EA, James JA, Hasler BP, Rosario-Rivera BL et al (2016). The role of non-rapid eye movement slow-wave activity in prefrontal metabolism across young and middle-aged adults. J Sleep Res 25: 296–306.
Wittchen HU, Krause P, Hofler M, Pittrow D, Winter S, Spiegel B et al (2001). [NISAS-2000: The ‘Nationwide Insomnia Screening and Awareness Study’. Prevalence and interventions in primary care]. Fortschr Med Orig 119: 9–19.
Youngstedt SD, Kripke DF (2004). Long sleep and mortality: rationale for sleep restriction. Sleep Med Rev 8: 159–174.
Yu H, Pardoll D, Jove R (2009). Stats in cancer inflammation and immunity: a leading role for stat3. Nat Rev Cancer 9: 798–809.
Zamarron F, Maceiras F, Mera A, Gomez-Reino JJ (2004). Effects of the first infliximab infusion on sleep and alertness in patients with active rheumatoid arthritis. Ann Rheum Dis 63: 88–90.
Zielinski MR, Dunbrasky DL, Taishi P, Souza G, Krueger JM (2013). Vagotomy attenuates brain cytokines and sleep induced by peripherally administered tumor necrosis factor-alpha and lipopolysaccharide in mice. Sleep 36: 1227–1238.
Acknowledgements
Work by MRI and MRO has been funded by the NIH. MRI and MRO declare no potential conflict of interest. Supported by grant R01-AG034588 from the National Institute of Aging, other grant support from the National Institutes of Health to MRI including R01-CA160245-01, R01 AG026364,R01-CA119159; R01-HL079955; R01 HL095799; and the Cousins Center for Psychoneuroimmunology.
Author information
Authors and Affiliations
Corresponding author
PowerPoint slides
Rights and permissions
About this article

Cite this article
Irwin, M., Opp, M. Sleep Health: Reciprocal Regulation of Sleep and Innate Immunity. Neuropsychopharmacol 42, 129–155 (2017). https://doi.org/10.1038/npp.2016.148
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2016.148
This article is cited by
-
Inverted U-shaped relationship between sleep duration and phenotypic age in US adults: a population-based study
Scientific Reports (2024)
-
Effect of CT-Guided Repeated Pulsed Radiofrequency on Controlling Acute/Subacute Zoster-Associated Pain: A Retrospective Cohort Study
Pain and Therapy (2024)
-
Characterization of sleep efficiency transitions in family caregivers
Journal of Behavioral Medicine (2024)
-
Opioid use disorder in two samples of the Lebanese population: scale validation and correlation with sleep and mood disorders
BMC Psychiatry (2023)
-
Heat of the night: sleep disturbance activates inflammatory mechanisms and induces pain in rheumatoid arthritis
Nature Reviews Rheumatology (2023)
