
Abstract
Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia (IBMPFD) is a dominant progressive disorder that maps to chromosome 9p21.1–p12. We investigated 13 families with IBMPFD linked to chromosome 9 using a candidate-gene approach. We found six missense mutations in the gene encoding valosin-containing protein (VCP, a member of the AAA-ATPase superfamily) exclusively in all 61 affected individuals. Haplotype analysis indicated that descent from two founders in two separate North American kindreds accounted for IBMPFD in ∼50% of affected families. VCP is associated with a variety of cellular activities, including cell cycle control, membrane fusion and the ubiquitin-proteasome degradation pathway. Identification of VCP as causing IBMPFD has important implications for other inclusion-body diseases, including myopathies, dementias and Paget disease of bone (PDB), as it may define a new common pathological ubiquitin-based pathway.
Similar content being viewed by others
Main
Hereditary inclusion body myopathy (IBM) associated with PDB and frontotemporal dementia (FTD), or IBMPFD, is a rare, complex and ultimately lethal autosomal dominant disorder (OMIM 605382; ref. 1). IBMPFD features adult-onset proximal and distal muscle weakness (clinically resembling limb girdle muscular dystrophy), early-onset PDB in most cases and 'premature' FTD2. Although the disorder was mapped to chromosome 9p21–p12, the genetic basis was not known.
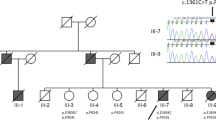
Within 13 families (12 from the United States and 1 from Canada, Fig. 1 and Supplementary Fig. 1 online), 82% of individuals had myopathy, 49% had PDB and 30% had early-onset FTD. The mean age of presentation was 42 years for both IBM and PDB, whereas FTD typically presented at age 53 years. In IBMPFD myopathic muscle and PDB osteoclasts, inclusions appear similar, suggestive of disruptions in the same pathological pathway.

Squares indicate males, and circles indicate females. Arrows indicate probands, and symbols with a slash indicate deceased family members. The filled upper right quadrant indicates IBM, filled lower right quadrant indicates PDB, and filled lower left quadrant indicates FTD. Only clinically diagnosed family members are shown, without ages, to preserve confidentiality.
Haplotype analysis of the families with IBMPFD identified two ancestral, disease-associated haplotypes, distinguishing families 1, 3, 7 and 16 (group A) from families 2 and 5 (group B), both groups of Northern European ancestry (Table 1). The predominant IBMPFD haplotype of group A includes a core haplotype flanked by D9S1118 and D9S234 (6.47 cM, 35.1 Mb), probably transmitted from a shared ancestor (founder). It is not known whether this individual emigrated to the US or the VCP mutation originated in the US and then radiated.
Using a candidate-gene approach, we first excluded GNE (UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase), which causes IBM2 or Nonaka myopathy and sialuria3,4, and then several additional genes5. We then identified six missense mutations (Table 1 and Supplementary Fig. 2 online) in VCP (also called CDC48 or p97)6. Families 1, 3, 4, 7, 10, 15 and 16 share the nucleotide substitution 464G→A (resulting in the amino acid substitution R155H) in exon 5; family 11 has the substitution 464G→C (R155P). Families 2 and 5 have an alteration at the first base of the same codon, 463C→T (R155C). Family 6 has the transition mutation 695C→A (A232E) in exon 6. Family 9 has the base change 283C→G (R95G) in exon 3, whereas family 13 has the change 572G→C (R191Q) in exon 5. Group A shares the substitution 464G→A (R155H), and group B shares the substitution 463C→T (R155C). Mutations in families 4, 10 and 15 (who have the same VCP mutation as group A but unique haplotypes) probably arose independently from those observed in group A. Families 6, 9, 11 and 13 do not share haplotypes and their VCP mutations are unique and must have arisen independently. Nevertheless, 10 of the 13 families with IBMPFD have an amino acid change at codon 155 in VCP, which seems, therefore, to be a mutation hot spot. Identification of six distinct cosegregating missense mutations in VCP identifies this as the genetic basis for IBMPFD.
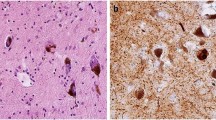
Immunohistochemical analysis of normal muscle sections with a polyclonal antibody to VCP showed staining of endomysial vessels, lipofuscin accumulations and, to a mild degree, muscle fiber cytoplasm (Fig. 2a). In sections of muscle with sporadic inclusion body myositis (s-IBM), however, VCP staining was localized to debris in inclusions and vacuoles (Fig. 2b). There was considerable staining of VCP in inflammatory cells at the focal invasion sites of muscle fibers (Fig. 2c). VCP was upregulated in regenerating muscle fibers with s-IBM (Fig. 2d). In muscles with IBMPFD, VCP was localized in large or small rounded aggregates in scattered muscle fibers (Fig. 2e,f), including those with no clear vacuoles or other morphological changes. Thus, VCP is commonly present in aggregates from muscle with IBMPFD and s-IBM, although the predominant pattern of localization for VCP differs between IBMPFD and s-IBM.

(a) Normal muscle. VCP is prominently located in small endomysial capillaries (right arrow). In muscle fibers, VCP accumulates with lipofuscin granules (left arrow) at the periphery and more diffusely, at low levels, in the cytoplasm. (b) Muscle with s-IBM. VCP is present in material in a vacuole (arrow) and in small accumulations in the muscle fiber cytoplasm. (c) Muscle with s-IBM. VCP is strongly stained in endomysial inflammatory cells surrounding a muscle fiber. (d) Muscle with s-IBM. VCP is upregulated in regenerating muscle fibers. (e) Muscle with IBMPFD. Large focal inclusion (arrow) in muscle fiber contains VCP. (f) Muscle with IBMPFD. Multiple small foci are present within a muscle fiber. Magnification: 540×.
Mutations of VCP in families 1–7, 9–11, 13, 15 and 16 cluster in the N-terminal CDC48 domain (Fig. 3a), which is involved in ubiquitin binding7,8. This highly structured domain forms two distinct regions9. VCP is conserved among species, and the amino acid residues mutated in IBMPFD are conserved in the higher mammals (Fig. 3b). VCP forms a homohexamer where the D1/D2 domains bind in a head-to-tail ring9, allowing the N-terminal domain to undergo conformational changes without affecting the stability of the homohexamer ring structure. VCP missense mutations causing IBMPFD disrupt the double ψ barrel (R95G in family 9), the four-stranded β barrel (R155C, R155H and R155P in families 1–5, 7, 10, 11 and 15) or the flexible linker (R191Q in family 13). Hence, the affected ubiquitin-binding domain may impair N-terminal domain binding of specific partner proteins. The mutation found in family 6 (A232E) affects the α5 helix of the a/b subdomain of the first AAA-ATPase domain (D1) and is potentially more deleterious because the D1 domain provides the protein's main catalytic activity essential for hexamer formation10. In fact, affected individuals in family 6 have fractures and PDB at an earlier age and their myopathy seems especially aggressive.

(a) Functional domains and mutations of VCP. Arrows indicate the locations of mutations relative to the exon-intron structure, where the exons are numbered 1–17. The relative positions of the N-terminal domain (CDC48, gray and cyan), flexible linker (L1, green), first AAA-ATPase domain (D1, blue), linker region (L2, dark grey), second AAA-ATPase domain (D2, dark blue) and C-terminal domain (black) are indicated, and the 5′ and 3′ untranslated regions are shown in white. (b) Species conservation of amino acid residues mutated in IBMPFD, with identical residues highlighted in red.
VCP has been associated with several essential cell protein pathways11: cell cycle, homotypic membrane fusion, nuclear envelope reconstruction, postmitotic Golgi reassembly, DNA damage response, suppressor of apoptosis and ubiquitin-dependent protein degradation12,13,14,15,16,17,18. VCP also binds to expanded polyglutamine (poly-Q) protein aggregates18,19. The poly-Q binding domain of human VCP maps to amino acid residues 142–200, which encompasses a region of the N-domain and linker (N domain to D1) that contains two of the mutations we identified18. A fruit fly VCP (ter94) loss-of-function mutant has been identified as a dominant suppressor of expanded poly-Q–induced neuronal degeneration20. The suppressive effects of the loss-of-function mutant did not seem to result from inhibition of poly-Q aggregate formation but rather from the degree of loss of VCP function. This suggests that a gene dosage response for VCP expression is essential to its function in expanded poly-Q–induced neuronal degeneration. In support of this idea, transgenic fruit flies in which VCP levels were elevated experienced severe apoptotic cell death, whereas homozygous VCP loss-of-function mutants were embryonic lethal20.
VCP is essential in the cell cycle and apoptosis pathways, neither of which seems to be disrupted in IBMPFD, as affected individuals are obviously viable. Clues about the nature of the mutations we identified in VCP can be drawn from pathways that have been implicated in other aggresome-associated degenerative disorders, which all involve protein quality control and the ubiquitin protein degradation pathways21,22,23,24. A number of independent studies support the fact that disruption of a specific function of VCP leads to inclusion body formation. First, experiments identifying the involvement of VCP in endoplasmic reticulum–associated degradation showed that dysfunction of VCP caused vacuole and inclusion body formation, ultimately leading to cell death18,19,25. Second, VCP interacts directly with polyubiquitinated proteins18,19,20. Third, VCP colocalizes with ubiquitin-containing nuclear inclusions in the cerebral cortex in a number of neuronal degenerative disorders involving protein quality control and the ubiquitin protein degradation pathways, such as Huntington, Alzheimer, Creutzfeldt-Jakob and Parkinson disease (in particular the Lewy bodies), as well as in motor neuron disease with dementia26. Fourth, mutations in the ubiquitin-binding domain of sequestosome 1 (SQSTM1, also called p62; refs. 27,28) cause autosomal dominant PDB (PDB3). We propose that mutations in VCP, as in SQSTM1, cause PDB by compromising ubiquitin-binding and target similar cellular pathways or proteins. Furthermore, p62 colocalizes with inclusion bodies in a number of degenerative disorders22,23. Thus, IBMPFD is probably a new member of the family of aggresome-associated disorders, and mutations in VCP may represent a new link in the pathway that leads to aggresome formation. Because IBMPFD is a dominant progressive syndrome, the mutations we identified are probably relatively subtle and aging, oxidative stress and endoplasmic reticulum stress probably define a threshold at which the IBMPFD phenotype becomes manifest. Rather than the mutations disrupting a normal function of VCP, they may add new, toxic functions that result in new VCP actions. Alternatively, the mutations could be dominant negative and disrupt normal hexamer formation of VCP.
In 13 families with IBMPFD, only four different amino acid residues (three in the N-terminal domain and one in the D1 domain) of VCP are mutated, suggesting either that there is a mutational hot spot in the N-domain or that VCP has such tight operational constraints that other types of mutation elsewhere are lethal. Indeed, homozygous loss-of-function fruit fly mutants are embryonic lethal20 and could explain the lack of a knockout mouse model for VCP. Our current findings identify a new and crucial role for VCP in aggresome disease, particularly in the seemingly unrelated tissues affected in IBMPFD, and indicate that VCP has more specialized functions still to be characterized.
Methods
Clinical evaluations.
We obtained written consent from each subject and approval for the study from the Springfield, Illinois Committee for Research Involving Human Subjects and from Children's Hospital, Boston, Massachusetts. Volunteers were all more than 18 years of age because IBMPFD typically manifests in adults. The IBMPFD muscle phenotype is of variable severity, and mild asymmetry characterizes the muscle weakness of IBMPFD1,2. Myopathic features include variation in muscle fiber size, mildly increased endomysial connective tissue and large focal regions of 'myopathic grouping' commonly seen in hereditary IBM2. Rimmed vacuolar inclusions were noted in ∼35% of muscle biopsy specimens analyzed from the 13 families. Electron microscopy of biopsy samples from individuals with IBMPFD showed atrophic and vacuolated muscle fibers containing abundant nuclear and cytoplasmic, paired helical filaments with congophilia, accumulations of phosphorylated tau, apolipoprotein E and excessive β-amyloid precursor protein epitopes1,2. Similarly, electron microscopy of PDB osteoclasts from four affected individuals with IBMPFD in family 11 identified seemingly characteristic nuclear and cytoplasmic paired helical filaments inclusions29.
Chromosomal mapping.
We extracted peripheral blood DNA using the PureGene DNA isolation kit (Gentra Systems). IBMPFD linkage to chromosome 9p21.1–p12 was known in four families2 and confirmed in the nine new kindreds. We constructed the disease-associated haplotype for each family to identify the critical locus2.
Assessments of candidate genes.
Our candidate approach involved genes prioritized and selected by their expression patterns and putative functions. We identified sequences provided by the Human Genome Project that contained Genethon markers that mapped to the disease-associated region. We then assessed each sequence using the National Center for Biotechnology Information BLAST search program to determine the exon-intron structures of candidate genes including VCP.
Mutation analysis of VCP.
Mutation analysis of VCP initially involved two affected individuals from each of the 13 families. Unaffected individuals and unrelated relatives served as controls. We designed PCR primers for genomic DNA to include at least 50 bp of intron sequence, from either side of the exon, for all 17 exons. Sequences longer than 1,000 bp were divided into multiple, overlapping segments for amplification. We gel-purified the PCR products using the Gel Extraction Kit (Qiagen) and sequenced them with an ABI 377 sequencer, using a dRhodamine terminator cycle sequencing kit (Applied BioSystems). We compared sequence and trace files using Lasergene 99 software (DNAStar). We then screened >180 additional control chromosomes by denaturing high-performance liquid chromatography (Transgenomic) and by restriction digests for any base changes in exons 3, 5 and 6, where mutations were identified, to rule out the possibility that the base changes were common polymorphisms. No base changes were detected in these control chromosomes.
Cosegregation studies.
We used restriction-site mapping from PCR-amplified exons to confirm that the mutations cosegregated with disease in families carrying the substitutions 695C→A (family 6), which destroys an MfeI site; 283C→G (family 9), which destroys an RSAI restriction site; and 464C→T (family 11), which creates a BSSKI restriction site. We confirmed cosegregation for the substitutions 463C→T, 464G→A and 572G→C using denaturing high-performance liquid chromatography in a blinded study of 79 individuals from seven families.
Immunohistochemistry.
To determine the presence of VCP in postmortem muscle sections from unaffected individuals, individuals with IBM and individuals with IBMPFD, we subjected sections to immunohistochemistry with polyclonal antibody to VCP as described previously2. We detected the immune reactivity by light microscopy using horseradish peroxidase.
URL.
The National Center for Biotechnology Information BLAST search program is available at http://www.ncbi.nlm.nih.gov/BLAST/.
GenBank accession number.
VCP, NM_007126.
Note: Supplementary information is available on the Nature Genetics website.
Accession codes
References
Kimonis, V.E. et al. Clinical and molecular studies in a unique family with autosomal dominant limb-girdle muscular dystrophy and Paget disease of bone. Genet. Med. 2, 232–241 (2000).
Kovach, M.J. et al. Clinical delineation and localization to chromosome 9p13.3-p12 of a unique dominant disorder in four families: hereditary inclusion body myopathy, Paget disease of bone, and frontotemporal dementia. Mol. Genet. Metab. 74, 458–475 (2001).
Eisenberg, I. et al. The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathy. Nat. Genet. 29, 83–87 (2001).
Seppala, R., Lehto, V.P. & Gahl, W.A. Mutations in the human UDP-N-acetylglucosamine 2-epimerase gene define the disease sialuria and the allosteric site of the enzyme. Am. J. Hum. Genet. 64, 1563–1569 (1999).
Watts, G.D.J., Thorne, M., Kovach, M.J., Pestronk, A. & Kimonis, V.E. Clinical and Genetic Heterogeneity in Chromosome 9p associated Hereditary Inclusion Body Myopathy: Exclusion of GNE and three other candidate genes. Neuromuscul. Disord. 13, 559–567 (2003).
Beyer, A. Sequence analysis of the AAA protein family. Protein Sci. 6, 2043–2058 (1997).
Dai, R.M. & Li, C.C. Valosin-containing protein is a multi-ubiquitin chain-targeting factor required in ubiquitin-proteasome degradation. Nat. Cell Biol. 3, 740–744 (2001).
Rape, M. et al. Mobilization of processed, membrane-tethered SPT23 transcription factor by CDC48(UFD1/NPL4), a ubiquitin-selective chaperone. Cell 107, 667–677 (2001).
Zhang, X. et al. Structure of the AAA ATPase p97. Mol. Cell 6, 1473–1484 (2000).
Wang, Q., Song, C. & Li, C.C. Hexamerization of p97-VCP is promoted by ATP binding to the D1 domain and required for ATPase and biological activities. Biochem. Biophys. Res. Commun. 300, 253–260 (2003).
Wang, Q., Song, C. & Li, C.C. Molecular perspectives on p97-VCP: progress in understanding its structure and diverse biological function. J. Struct. Biol. advance online published, 19 December 2003 (doi:10.1016/j.jsb.2003.11.014).
Hetzer, M. et al. Distinct AAA-ATPase p97 complexes function in discrete steps of nuclear assembly. Nat. Cell Biol. 3, 1086–1091 (2001).
Rabinovich, E., Kerem, A., Frohlich, K.U., Diamant, N. & Bar-Nun, S. AAA-ATPase p97/Cdc48p, a cytosolic chaperone required for endoplasmic reticulum-associated protein degradation. Mol. Cell. Biol. 22, 626–634 (2002).
Rabouille, C. et al. Syntaxin 5 is a common component of the NSF- and p97-mediated reassembly pathways of Golgi cisternae from mitotic Golgi fragments in vitro. Cell 92, 603–610 (1998).
Kondo, H. et al. p47 is a cofactor for p97-mediated membrane. Nature 388, 75–78 (1997).
Meyer, H.H., Shorter, J.G., Seemann, J., Pappin, D. & Warren, G. A complex of mammalian ufd1 and npl4 links the ubiquitin and nuclear transport pathways. EMBO J. 19, 2181–2192 (2000).
Jarosch, E., Geiss-Friedlander, R., Meusser, B., Walter, J. & Sommer, T. Protein dislocation from the endoplasmic reticulum—pulling out the suspect. Traffic 3, 530–536 (2002).
Hirabayashi, M. et al. VCP/p97 in abnormal protein aggregates, cytoplasmic vacuoles, and cell death, phenotypes relevant to neurodegeneration. Cell Death Differ. 8, 977–984 (2001).
Kobayashi, T., Tanaka, K., Inoue, K. & Kakizuka, A. Functional ATPase activity of p97/valosin-containing protein (VCP) is required for the quality control of endoplasmic reticulum in neuronally differentiated mammalian PC12 cells. J. Biol. Chem. 277, 47358–47365 (2002).
Higashiyama, H. et al. Identification of ter94, Drosophila VCP, as a modulator of polyglutamine-induced neurodegeneration. Cell Death Differ. 9, 264–273 (2002).
Layfield, R., Alban, A., Mayer, R.J. & Lowe, J. The ubiquitin protein catabolic disorders. Neuropathol. Appl. Neurobiol. 27, 171–179 (2001).
Garcia-Mata, R., Gao, Y.S. & Sztul, E. Hassles with taking out the garbage: aggravating aggresomes. Traffic 3, 388–396 (2002).
Donaldson, K.M. et al. Ubiquitin-mediated sequestration of normal cellular proteins into polyglutamine aggregates P62 and the sequestosome, a novel mechanism for protein metabolism. Proc. Natl. Acad. Sci. USA 100, 8892–8897 (2003).
Wood, J.D., Beaujeux, T.P. & Shaw, P.J. Protein aggregation in motor neurone disorders. Neuropathol. Appl. Neurobiol. 29, 529–545 (2003).
Nagahama, M. et al. SVIP is a novel VCP/p97-interacting protein whose expression causes cell vacuolation. Mol. Biol. Cell 14, 262–273 (2003).
Mizuno, Y., Hori, S., Kakizuka, A. & Okamoto, K. Vacuole-creating protein in neurodegenerative diseases in humans. Neurosci. Lett. 343, 77–80 (2003).
Hocking, L.J. et al. Domain-specific mutations in sequestosome 1 (SQSTM1) cause familial and sporadic Paget's disease. Hum. Mol. Genet. 11, 2735–2739 (2002).
Laurin, N., Brown, J.P., Morissette, J. & Raymond, V. Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am. J. Hum. Genet. 70, 1582–1588 (2002).
Yabe, H., Singer, F.R., Tucker, W.S. Jr. & Mills, B.G. Paget-like inclusions in osteopetrosis and hereditary neuromuscular and skeletal disease. Eighth Annual Meeting of the American Society of Bone and Mineral Research, A221 (1986).
Acknowledgements
We thank the families and their physicians, in particular F. Singer, S. Markus, K. Boycott, B. Sundaram, S. Tucker, Z. Simmons, J. Towfighi, E. Neilan, C. Smith and G. Umberger, for their participation in and contribution to our research studies; C.C. Li for the VCP antibody; S. Banze and M. Geimer for molecular analyses; B. Waggoner for her preliminary work; and L. Kunkel, A. Beggs, E. Gussoni and M. Irons for their support. Funding of this study is from the US National Institutes of Health, the Muscular Dystrophy Association and the Paget Foundation, Children's Hospital Boston Equipment Grant, Shriners Hospitals for Children, the Barnes-Jewish Hospital Foundation and previously from the Excellence in Academic Medicine Program at Southern Illinois University School of Medicine.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
About this article
Cite this article
Watts, G., Wymer, J., Kovach, M. et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet 36, 377–381 (2004). https://doi.org/10.1038/ng1332
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng1332
This article is cited by
-
UBXN1 maintains ER proteostasis and represses UPR activation by modulating translation
EMBO Reports (2024)
-
Amyotrophic lateral sclerosis: translating genetic discoveries into therapies
Nature Reviews Genetics (2023)
-
The AAA+ chaperone VCP disaggregates Tau fibrils and generates aggregate seeds in a cellular system
Nature Communications (2023)
-
Patients with sporadic FTLD exhibit similar increases in lysosomal proteins and storage material as patients with FTD due to GRN mutations
Acta Neuropathologica Communications (2023)
-
DUSP1 interacts with and dephosphorylates VCP to improve mitochondrial quality control against endotoxemia-induced myocardial dysfunction
Cellular and Molecular Life Sciences (2023)
