
Abstract
Ligand binding sites within proteins can interact by allosteric mechanisms to modulate binding affinities and control protein function. Here we present crystal structures of the regulator of K+ conductance (RCK) domain from a K+ channel, MthK, which reveal the structural basis of allosteric coupling between two Ca2+ regulatory sites within the domain. Comparison of RCK domain crystal structures in a range of conformations and with different numbers of regulatory Ca2+ ions bound, combined with complementary electrophysiological analysis of channel gating, suggests chemical interactions that are important for modulation of ligand binding and subsequent channel opening.
Similar content being viewed by others
Introduction
Regulator of K+ conductance (RCK) domains control the activity of a variety of K+ channels and transporters, including the prokaryotic TrkA/H K+ transport complex and the eukaryotic large-conductance calcium-activated (BK) channel, through binding of cytoplasmic ligands such as ATP, H+ and Ca2+ (refs 1, 2, 3, 4, 5, 6, 7, 8). Thus, RCK domains transduce ligand binding to gate transmembrane K+ flux in response to signalling events and cellular metabolism, in organisms ranging from bacteria to humans. In addition, binding of one ligand to an RCK domain can allosterically modulate binding of a second ligand, resulting in tuning of channel or transporter activity in response to cellular stimuli9,10,11. These allosteric interactions are key in generating a wide dynamic range for ligand activation of channels and transporters, in addition to linking multiple stimulus modalities to gate ion flux. Although allosteric interactions in the RCK domains have been described in functional terms, the structural bases for such interactions are yet unclear10,12,13.
Using electrophysiology and X-ray crystallography, we address the structural basis of allosteric interaction among Ca2+-binding sites in the MthK Ca2+-gated K+ channel. MthK is a prototypical K+ channel, whose crystal structure has provided insight towards mechanisms of channel gating by RCK domains4,9,10,14,15,16,17,18. In MthK, binding of cytoplasmic Ca2+ to a tethered octameric ring of RCK domains (the ‘gating ring’) leads to a series of conformational changes that facilitates channel opening and K+ conduction4,10,19,20,21. Here, by solving structures of mutant and wild-type (WT) RCK domains, we find that distinct Ca2+ activation sites near the amino and carboxy termini of the RCK domain (termed C1 and C3, respectively) are allosterically coupled to one another, to affect tuning of Ca2+ affinity and Ca2+-dependent channel activation. These results define a structural mechanism of allosteric modulation in a ligand-gated K+ channel, and provide a framework for understanding similar mechanisms in related RCK-containing channels and transporters.
Results
The E212Q mutation paradoxically enhances Ca2+ sensitivity
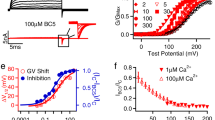
MthK contains three Ca2+-binding sites per RCK domain that facilitate channel activation (termed C1, C2 and C3; Fig. 1)16. Consistent with the activating role of Ca2+, we observe that charge neutralization of the Ca2+-coordinating side chains D184 and E210 at the Ca2+-binding site C1 leads to MthK channels with decreased Ca2+ sensitivity; the [Ca2+] yielding half-maximal channel open probability (EC50) was 2.2±0.1 mM for WT (n=5), 6.3±0.4 mM for D184N (n=5) and 4.0±0.3 mM for E210Q (n=6), all at pH 7.7 (throughout, EC50 values reported for each mutant are the means of EC50 values determined from individual patches±s.e.m.). However, neutralization of the Ca2+-coordinating side chain E212 at the same binding site yields channels with increased Ca2+ sensitivity (EC50=1.3±0.1 mM Ca2+ for E212Q, n=8, pH 7.7; Fig. 1f). Ca2+ activation of MthK is known to be sensitive to pH, and these mutant effects persisted at pH 6.5 (EC50=16±1.6 mM Ca2+ for WT, n=6; 30±2.0 mM Ca2+ for D184N, n=7; and 9.9±0.3 mM Ca2+ for E212Q, n=5; Fig. 1g); although increasing [H+] leads to an overall shift in EC50 toward higher [Ca2+], the E212Q mutant consistently requires less Ca2+ for activation than MthK WT.

(a) Structure of the MthK channel shown as surface rendering (PDB accession number 3RBZ). The pore (in the plasma membrane, represented by grey shading) and its contiguous RCK domains (at the cytoplasmic side) are coloured cyan; dashed cyan lines represent the unresolved pore–RCK linker residues. Associated RCK domains (which complete the gating ring complex) are coloured yellow. (b) Cα trace of the gating ring superimposed within its surface rendering as viewed from above the membrane; the pore has been removed for clarity. RCK domains are coloured as in a; Ca2+ ions are shown as green spheres. Locations of representative C1, C2 and C3 Ca2+ ions are indicated by circles. (c) Magnified view of the C1 Ca2+-binding site; dashed lines indicate coordination by oxygens from carboxylate side chains of D184, E210 and E212. (d) Representative single-channel current traces from WT and C1 mutant MthK channels, at indicated [Ca2+]i (left) or [Cd2+]i (right). D184N channels show reduced Ca2+ and Cd2+ sensitivity, but E212Q channels show enhanced sensitivity. These and subsequent current traces were recorded in symmetrical 200 mM KCl, Vm=−100 mV. (e) Activation of WT, D184N and E212Q channels by Ca2+ at pH 7.7 (WT, n=5; D184N, n=5; and E212Q, n=8). (f) Activation of WT, D184N and E212Q channels by Ca2+ at pH 6.5 (WT, n=6; D184N, n=7; and E212Q, n=5). (g) Activation of WT, D184N and E212Q channels by Cd2+ at pH 7.7 (WT, n=8; D184N, n=6; and E212Q, n=5). Data points indicate mean Po±s.e.m. at the indicated ligand concentration.
Aside from Ca2+, MthK channels can also be activated by Cd2+ with EC50 in the micromolar range, through binding of Cd2+ ions at the C1 and C3 activation sites21,22,23. Figure 1 illustrates that the MthK WT channel is activated by Cd2+ with an EC50 of 49.1±5.0 μM (n=8). The functional effects of the D184N and E212Q mutations on Cd2+ activation, indicated by the shift in EC50, were even stronger than their effects on Ca2+ activation. D184N channels were activated with an EC50 of 880±80 μM Cd2+ (n=6, an 18-fold increase in EC50 compared with that in WT), whereas E212Q channels were activated with an EC50 of 16.3±2.9 μM Cd2+ (n=5; a 3-fold decrease in EC50 compared with that in WT).
Charge-neutralizing mutation is a commonly used approach towards identifying Ca2+-coordinating side chains in proteins24,25,26,27. Thus, our initial goal in the present work was to learn the relation between these mutation effects and the Ca2+-activation mechanism in MthK, in part by solving crystal structures of binding-site mutants in the presence of Ca2+.
Effects of Ca2+ on RCK domain conformation
To gain insight towards the structures of the Ca2+-binding sites in the MthK RCK domain, we crystallized the RCK domain in the presence of Ca2+, in a crystal form that diffracted X-rays to 2.5 Å, in space group P3121 (Fig. 2). The structure was solved by molecular replacement and refined to Rwork/Rfree of 0.178/0.226 (Table 1). As strong electron density peaks were observed at each of the three Ca2+ activation sites, termed C1, C2 and C3, we refer to this structure as WT-C1C2C3. The Ca2+-bound RCK domains in this structure form a dimer, with each peptide chain consisting of an N-lobe, a domain-swapping helix-turn-helix motif and a C-lobe (Fig. 2a). In this context, it can be seen that the C1 site is contained entirely within a single N-lobe (Fig. 2b), whereas the Ca2+ ion of the C2 site forms an intersubunit bridge between the ‘turn’ of the helix-turn-helix motif and the C-lobe of adjacent subunits (Fig. 2c), and the Ca2+ ion of the C3 site also forms an intersubunit bridge between C-lobes of adjacent subunits (Fig. 2d).

(a) Schematic representation of the RCK domain dimer. Component subunits are coloured cyan and yellow; Ca2+ ions and water are represented by green and red spheres, respectively. Locations of the N-lobe, helix-turn-helix (HTH) and C-lobe are as indicated. Dashed boxes indicate locations of Ca2+ sites. (b–d) Ball and stick representations of Ca2+ sites C1 (b), C2 (c) and C3 (d). Ca2+ coordination is illustrated by dashed lines.
It is worth noting that the overall conformation of the WT-C1C2C3 structure recapitulates the RCK domain conformation within the gating ring of the Ca2+-bound, open MthK channel, although the WT-C1C2C3 structure did not itself crystallize as a gating ring, and the WT-C1C2C3 crystals are comprises the RCK domain only, not tethered to the MthK pore (Figure 3)4,16. The similarity between the WT-C1C2C3 structure and the RCK domains from the fully liganded open channel is thus consistent with the idea that the domain conformation depends primarily on Ca2+ occupancy at the activation sites, and is not strictly dependent on tethering to the MthK pore or formation of the gating ring complex.

Overlayed Cα traces of WT-C1C2C3 (yellow/cyan) and RCK domains from Ca2+-bound MthK channel (PDB accession number 3RBZ; orange/purple). The small overall root mean squared difference for these aligned structures (0.8 Å) suggests that they represent nearly identical conformations.
The WT-C1C2C3 structure defines previously unresolved details of ion coordination within the domain’s Ca2+ activation sites. For example, the Ca2+ ion at each site was surrounded by protein oxygens and ordered solvent molecules, exhibiting canonical bipyramidal coordination geometries (Fig. 2)16,28,29,30. Notably, the C2 Ca2+ ion is seen to be coordinated at the centre of a ring formed by side-chain oxygens from residue E248, the carbonyl oxygen from D245 and a water of hydration, with the capstones of each pyramid formed by a side-chain oxygen from E266 and the carbonyl oxygen from R241. Similarly, the C3 Ca2+ ion is coordinated at the centre of a ring formed by side-chain oxygens from residues D305 and E326, and one water of hydration, with a carbonyl oxygen from G290 and a second water of hydration forming the capstones of each pyramid. Finally, the C1 Ca2+ ion is at the centre of a ring formed by two oxygen atoms from the D184 side chain, two water molecules and one oxygen atom from the E212 side chain28.
As both D184 and E212 directly coordinate the Ca2+ ion at C1 in the context of the fully liganded RCK domain, one might predict that neutralization of D184 or E212 by mutation to N or Q, respectively, would weaken the electrostatic interaction with Ca2+ in both cases, and thus decrease Ca2+-dependent channel activation. Although the D184N mutation decreases Ca2+ activation, the E212Q mutation unexpectedly increases Ca2+ activation (Fig. 1). We next addressed the structural basis of these functional effects.
Crystal structures of gating mutants
To determine whether mutations at the C1 site affect only Ca2+ binding at C1 or, alternatively, have allosteric effects at other Ca2+-binding sites, we sought to compare the structures of WT and mutant RCK domains, crystallized under the same conditions as one another, to minimize the contribution of differences in pH or ionic strength to structural changes that might be observed. To find conditions that would allow such direct comparison of mutant and WT domain structures, we started with the crystallization buffer that yielded the WT-C1C2C3 structure and examined crystals formed with lower [Ca2+] to yield WT crystals in which not all Ca2+ sites were occupied. When [Ca2+] was lowered within the range of 20–300 mM CaCl2 in this crystallization buffer, WT RCK domains crystallized in space group P21 and contained a Ca2+ ion bound only at the C1 site. The C3 sites in this structure are in conformations that are different from the C3-bound structure (Fig. 4a,b compared with Fig. 2b,d). Although one cannot determine binding constants from these data, the observation that lowering [Ca2+] leads to dissociation of Ca2+ from C3 but not C1 is consistent with C1 being the higher-affinity site under these conditions. This structure, termed WT-C1 (formed in 200 mM Ca2+), was refined to 1.85 Å resolution with Rwork/Rfree of 0.211/0.253 (Table 1). The crystallization conditions for WT-C1 were then used as a template for crystallization of the D184N and E212Q mutant RCK domains.

(a,b) Structures of the C1 and C3 sites from the WT RCK domain crystallized with 0.2 M Ca2+, overlayed with 2Fo–Fc map contoured at 1.2σ (blue mesh) and 5σ (purple mesh). (c,d) Structures of the same regions from the D184N mutant RCK domain, overlayed with 2Fo–Fc map contoured at 1.2σ (blue mesh) and Fo–Fc map contoured at 4σ (positive peaks shown as green mesh), indicating the absence of significant unmodelled electron density at the C1 site. (e) Structure of the C1 site from the E212Q mutant RCK domain crystallized under the same conditions as WT-C1, overlayed with 2Fo–Fc map contoured at 1.5σ (blue mesh). (f) Structure of the C3 site from the E212Q mutant RCK domain, overlayed with 2Fo–Fc map contoured at 1.5σ (blue mesh) and 5σ (purple mesh). Maps were calculated using data to 1.85 Å (a,b), 2.4 Å (c,d) and 3.0 Å resolution (e,f).
The D184N RCK domain crystallized in space group P6522 and the structure was refined to 2.4 Å resolution with Rwork/Rfree of 0.197/0.247 (Table 1). In this structure, no electron density that could be modelled as a Ca2+ ion was detectable at C1 (Fig. 4c). However, interestingly, density corresponding to Ca2+ ions was clearly observed at C3; the resolution of these data was sufficient to resolve ordered solvent surrounding the Ca2+ ions at C3, recapitulating the coordination geometry observed in the WT-C1C2C3 structure (Fig. 4d, compared with Fig. 2d). As the [Ca2+] in the D184N crystallization buffer was the same as that in the WT-C1 buffer, these results suggest that the D184N mutation has altered the relative affinities of the C1 and C3 sites for Ca2+. This observation further suggests that these two Ca2+ sites are allosterically coupled to one another, that is, the D184N mutation, which directly alters Ca2+ binding at the C1 site, also effects Ca2+ binding at the physically distinct C3 site.
To gain further insight towards the allosteric relation between C1 and C3, we crystallized the E212Q RCK domain. This mutant also crystallized in space group P6522 and the structure was refined to 3.0 Å resolution with Rwork/Rfree of 0.217/0.263. In contrast to D184N, the E212Q mutant did contain electron density at the C1 and C3 site, modelled as Ca2+ (Figs 4e,f and 5). Interestingly, however, Ca2+ coordination at the C1 site of the E212Q mutant is different from the configuration of the C1 site in the WT-C1 or WT-C1C2C3 structures. At the WT C1 site, the E212 side chain appears to directly coordinate Ca2+, with a mean Ca2+–Oε distance of ~2.5 Å (Figs 1c and 4a)16,28. In contrast, the nearest Oε/Nε atoms from Q212 are further from Ca2+ than the canonical coordination distance (mean distance=3.3 Å; Fig. 4e), suggesting that Q212 does not participate in direct coordination of the C1 Ca2+ ion. The distance between Ca2+ and the other key oxygen ligands, from the D184 and E210 side chains, are similar to those observed in the WT-C1 or WT-C1C2C3 structures (Figs 2b and 4a; mean distances for E212Q structure were 2.6 Å for D184/Oδ and 2.5 Å for E210/Oε). Thus, the position of the Q212 side chain in the E212Q mutant may be less constrained by Ca2+ coordination than the E212 side chain in the WT RCK domain. As the E212 side chain would similarly be unconstrained by Ca2+ in the D184N mutant, Ca2+ coordination by the E212 side chain may be important in allosteric coupling between the C1 and C3 Ca2+-binding sites.

To further validate modelling of Ca2+ ions at the C1 site in the E212Q structure, we calculated omit maps to minimize bias arising from model-derived phase information. The complete structure of the E212Q C1 site is shown in ball and stick representations, with Ca2+ ion as green spheres. The omit map (purple mesh, contoured at 1.5σ) was calculated by omitting Ca2+ ions and surrounding side chains (D184, E210 and Q212) from this structure, then performing simulated annealing refinement to eliminate the contribution of the omitted atoms in the calculation of phase information. The position of the electron density peak in the omit map that coincides with the position of the Ca2+ ion in the complete structure is thus consistent with the presence of Ca2+ at the site.
The effect of mutation on Ca2+-binding at sites C1 and C3 observed in the D184N and E212Q crystal structures suggest a mechanism underlying the functional effects of these mutations on Ca2+-dependent gating of MthK. Ca2+ binding at C1, which appears to be the higher-affinity site in the WT RCK domain, makes Ca2+ binding at C3 relatively unfavourable (Fig. 4a,b). The D184N mutation, which abrogates Ca2+ binding at C1, makes Ca2+ binding at C3 more favourable than it is in WT under the same conditions; however, the apparent loss of Ca2+ binding at C1 results in an overall decreased Ca2+ sensitivity of the D184N mutant channel (Figs 1d–g and 4c). As the E212Q mutation also makes Ca2+ binding at C3 more favourable than it is in WT, but does not eliminate Ca2+ binding at C1, Ca2+ activation of E212Q channels is enhanced over that of the WT (Figs 1d–g and 3e,f).
Structural basis of allosteric coupling
We reasoned that if C1 and C3 are allosterically coupled to one another, then they must be linked through a series of chemical bonds such that Ca2+ binding at C1 raises the energy barrier for Ca2+ binding at C3. Thus, we hypothesized that interactions (for example, salt bridges or hydrogen bonds) in the RCK domain that are correlated with occupancy or vacancy of C3 may determine part of the physical link between these sites and may underlie their allosteric modulation. To test this hypothesis, we systematically compared the structures of WT-C1, WT-C1C2C3, D184N and E212Q with the goal of identifying specific chemical bonds that are correlated with the presence or absence of Ca2+ at C3.
Among the potential polar and electrostatic interactions present in the MthK RCK domain, the side chains of E133 and E259 in WT-C1 had Oε–Oε distances of 2.8 Å, consistent with a carboxyl–carboxylate hydrogen bond (that is, <3.2 Å; Fig. 6). In contrast, these Oε–Oε distances were much greater in the WT-C1C2C3, D184N and E212Q structures (WT-C1C2C3, 6.2 Å; D184N, 7.3 Å; and E212Q, 8.3 Å; Figs 6 and 7). To test further whether the hydrogen bonding observed with WT-C1 was present or absent in previous MthK structures, we examined structures of the RCK domain crystallized with 0 and 20 mM Ca2+ (PDB accession numbers 2AEJ and 2AEF, solved to 2.1 and 1.7 Å resolution, respectively)28. In these structures, minimum Oε–Oε distances for E133–E259 were 3.1 Å and 2.8 Å for 0 and 20 mM Ca2+, respectively (Fig. 7).

(a) Aligned Cα traces of WT-C1 (dark yellow/grey) and WT-C1C2C3 (light yellow/cyan) illustrating apparent movement of the C-lobe (inside black dashed box) and E259 side chain (inside blue dashed box) relative to other regions. (b) Expanded view of the large boxed region in a, with magenta dashed arrows indicating the direction of apparent Ca2+-dependent movement. (c) Expanded view of the smaller boxed region in a, with magenta dashed arrows indicating the direction of the apparent movement of the E259 side chain, relative to E133; E259* indicates the E259 side chain from WT-C1C2C3, with Ca2+ bound at C3. (d) Representative single-channel current traces of MthK WT, E259A and E212Q/E259A mutant channels over a range of [Ca2+]. Channels containing the E259A mutation alone or in combination with E212Q show enhanced Ca2+ sensitivity compared with WT. (e) Po versus [Ca2+] for WT and mutant channels. Data points indicate mean Po±s.e.m. Activation curves for E212Q (n=8), E259A (n=5) and E212Q/E259A channels (n=5) are each shifted towards lower [Ca2+] relative to WT, consistent with decreased inhibitory coupling.

(a–g) Cα traces of the indicated structures with E259 and E133 side chains shown as ball and stick representations. The minimal distances (in Å) between E259 and E133 Oε atoms are indicated by dashed magenta lines. The shortest distances among these, consistent with a hydrogen bond length (<3.2 Å), are associated with structures in which no Ca2+ is bound at the C3 site (a–c). In contrast, longer distances are associated with structures of WT (d,e) or mutant RCK domains (f,g) in conditions where Ca2+ ions are bound at the C3 site. Structures in a and b (PDB accession numbers 2AEJ and 2AEF, respectively) are from ref. 28; structure in d (PDB accession number 3RBZ) is from ref. 16.
The close proximity between the E133 and E259 side chains in the WT-C1 structure and the previous structures with 0 and 20 mM Ca2+ predicts that these side chains are at least partially protonated; if these side chains were deprotonated, both would carry negative charges, and thus repel one another. Although the WT-C1C2C3, D184N and E212Q structures each show greater Oε–Oε distances for these side chains compared with WT-C1, it should be noted that all of these crystals were grown at the same pH. Thus, the differences in conformation are not likely to arise from differences in side-chain protonation. Together, these results suggest that the E133–E259 interaction is correlated with the absence of Ca2+ occupancy at C3; however, it is not clear from these structural observations alone whether this interaction is related to channel gating.
We next tested whether the E133–E259 interaction affects channel gating, by assaying the Ca2+-activation properties of the E259A mutant channels, designed to constitutively disrupt the carboxyl–carboxylate interaction. We observe that the E259A-gating phenotype was very similar to E212Q, with EC50 values that were shifted towards lower [Ca2+] compared with WT (mean EC50=1.4±0.1 mM Ca2+, n=5; Fig. 6d,e). This electrophysiological result is consistent with the idea that the E259 side chain contributes to inhibitory coupling between C1 and C3.
If E212 and E259 each contribute to allosteric coupling between C1 and C3, then one might predict that the combined effects of the E212Q and E259A mutations on Ca2+ activation of the channel would be energetically non-additive31. To test this, we assayed Ca2+ activation of the E212Q/E259A double mutant and found that the EC50 of this double mutant (1.4±0.2 mM Ca2+, n=5) was not significantly different from either the E212Q or E259A single mutants (Fig. 6e). Although we cannot yet rule out that other modulatory interactions exist, the observation that the E212Q/E259A double mutant is effectively indistinguishable from the single mutants suggests that either disruption of the direct E212-Ca2+ bond or the E259–E133 hydrogen bond is sufficient to disrupt allosteric communication between these sites.
Discussion
Here we have used electrophysiological and crystallographic analysis of MthK to dissect potential mechanisms underlying activation and modulation of RCK domains. We observe principally that two physically distinct Ca2+-binding sites on the RCK domain are structurally and energetically coupled to one another, and we further define a key interaction that mediates this allosteric coupling to tune Ca2+ sensitivity of the channel.
A working hypothesis for the allosteric coupling mechanism is illustrated in Fig. 8. In this scheme, with Ca2+-binding sites empty (Fig. 8a), the RCK domains may exist in a range of conformations, as observed in structures of the unliganded MthK gating ring17,21. Ca2+ binding at the C1 sites (in the N-lobes) would then lead to stabilization of a partially activated conformation16,21 (Fig. 8b). Structures of WT and mutant RCK domains that display differences in Ca2+ occupancy at the C1 and C3 sites (and different overall conformations) further suggest that movements of the N-lobe and C-lobe are coupled to one another, attributable in part to an apparent carboxyl–carboxylate hydrogen bond between E133 and E259 (Figs 6 and 7). With the E133–E259 interaction intact, the Ca2+-bound conformation of the N-lobe is incompatible with Ca2+ binding to the C3 sites (Figs 6 and 7). Thus, it appears that Ca2+ binding at C3 requires either disruption of the E133–E259 interaction (Fig. 8c), as with the E259A mutation, or disruption of Ca2+ coordination at C1, as with the D184N and E212Q mutations (Figs 4 and 5). Ca2+ occupancy at both the C1 and C3 sites is required for complete activation of the RCK domain and maximal stabilization of channel opening.

(a) With Ca2+-binding sites empty, the RCK domains may exist in a range of conformations (represented by dashed magenta arrows). Movements of the N-lobe and C-lobe are coupled to one another, due in part to the E133–E259 carboxyl–carboxylate bond (blue triangles, highlighted in red). Ca2+ ions (green spheres) initially bind to the C1 sites in the N-lobe of each domain. (b) With Ca2+ bound at the C1 sites, a partially activated conformation is stabilized, but Ca2+ binding to the C3 sites is blunted, due in part to the E133–E259 interaction. (c) Increasing [Ca2+] further leads to Ca2+ binding to the C3 sites, which bridge the C-lobes of adjacent RCK domains. The conformational change associated with C3 binding involves movements of E259 and the C-lobe (represented by magenta highlighting and dashed arrows, respectively).
An inhibitory interaction between two Ca2+-binding sites in the same protein may represent a structural mechanism that underlies ‘tuning’ of the protein’s Ca2+ sensitivity. In the case of MthK, crystallographic structures indicate that the C1 site, located within the N-lobe of each RCK domain, undergoes relatively subtle structural changes upon Ca2+ binding and appears to bind Ca2+ with higher relative affinity than the C3 site (Fig. 4a,b)16,28. Thus, in conditions where Ca2+ binding at C1 inhibits binding at C3, a Ca2+ signal may lead to Ca2+ binding at the C1 site and, in turn, may be sufficient for partial activation of the channel21. On the other hand, ionic or metabolic conditions that weaken inhibitory coupling would result in increased Ca2+ affinity at C3, and thus lead to greater channel activation in response to an equivalent Ca2+ signal. The carboxyl–carboxylate bond between E133 and E259 would require at least partial protonation of these side chains; thus, with decreased [H+] (that is, elevated pH), one would expect these side chains to become deprotonated and thus negatively charged. If both E133 and E259 were negatively charged, they would electrostatically repel one another, and the inhibitory coupling would be weakened. Ca2+ activation of MthK is known to be greater with decreased [H+]; thus, this mechanism could account for a component of the H+ sensitivity of MthK gating, although other mechanisms may be involved10,19,21.
Similar to MthK, the eukaryotic BK channel also contains RCK domains that are modulated by multiple, separate Ca2+-binding sites13,32,33. Functional analyses of the BK channel gating indicates that these separate binding sites can interact energetically34; however, the structural basis of these interactions is not yet clear. As the BK channels are thought to be regulated by a number of potential physiological modulators, including H+, haem, carbon monoxide, nitric oxide and phosphorylation35, it will be important to apply structural analysis to better understand mechanisms underlying gating and modulation in these and other families of ion channels.
Methods
Channel purification and reconstitution
The full-length MthK channel, containing a C-terminal thrombin cleavage site followed by a 6xHis-tag, was expressed in Escherichia coli XL-1 Blue cells on induction with 0.4 mM IPTG (isopropyl β-D-1-thiogalactopyranoside)10. Bacteria were collected and resuspended in 20 mM Tris, 100 mM KCl, pH 7.6 (Buffer A), and lysed by sonication in the presence of phenylmethylsulphonyl fluoride and a protease inhibitor cocktail (Complete EDTA-free, Roche). The protein was solubilized by 2 h incubation in Buffer A with 50 mM decyl maltoside (DM), followed by centrifugation at 30,500 g for 45 min. The supernatant was loaded onto a Co2+-charged HiTrap metal affinity column (GE Healthcare), washed with 20 mM imidazole and 5 mM DM in Buffer A, and the channel protein was eluted using 400 mM imidazole and 5 mM DM in Buffer A. The 6xHis-tag was cleaved immediately after elution by incubating with 2.0 units of thrombin/3.0 mg eluted protein for 2 h at room temperature. Protein was concentrated using a centrifugal filtration unit (50 kDa molecular weight cut-off value) and further purified on a Superdex 200 gel filtration column, then concentrated again to 5 mg ml−1. Channels were reconstituted into liposomes composed of E. coli lipids (Avanti)36, and the proteoliposomes were rapidly frozen in liquid N2 and stored at −80 °C until use. Mutations were generated using the QuickChange Site-directed Mutagenesis Kit (Stratagene) and confirmed by DNA sequencing.
Electrophysiology
Recordings were obtained using planar lipid bilayers of POPE:POPG (3:1), in a horizontal bilayer chamber, at 22–24 °C. Solution in the cis (top) chamber contained 200 mM KCl and 10 mM HEPES, pH 7.0; solution in the trans (bottom) chamber contained 200 mM KCl, with either 10 mM HEPES (to buffer solutions to 7.7) or 10 mM 2-(N-morpholino)ethanesulfonic acid (MES) (to buffer solutions to pH 6.5), and CaCl2 or CdCl2 at the indicated concentrations. Recordings analysed for this work were obtained at −100 mV transmembrane voltage, although currents through each bilayer were observed at additional voltages to confirm the reversal potential and other MthK channel properties37. Within each bilayer, multiple solution changes were performed using a gravity-fed perfusion system, and to ensure completeness of solution changes the trans chamber was washed with a minimum of ten chamber volumes of solution before recording under a given set of conditions.
Currents were amplified using a Dagan PC-ONE patch-clamp amplifier with low-pass filtering to give a final effective filtering of 1 kHz (dead time of 0.18 ms) and were sampled by computer at a rate of 10 kHz. Currents were analysed by measuring durations of channel openings and closings at each current level by 50% threshold analysis, using pClamp9. These measurements were used to quantify channel activity as NPo= , where i is the open level and Pi is the per cent of time at that level. The mean channel open probability (Po) is then calculated by dividing NPo times N, which is the number of active channels in the bilayer, determined by recording under conditions where maximal channel opening is observed. Data are presented as mean±s.e.m.
, where i is the open level and Pi is the per cent of time at that level. The mean channel open probability (Po) is then calculated by dividing NPo times N, which is the number of active channels in the bilayer, determined by recording under conditions where maximal channel opening is observed. Data are presented as mean±s.e.m.
EC50 values were estimated by fitting Po versus [Ca2+] or Po versus [Cd2+] data for individual bilayer patches using: Po=Pomax/(1+([X2+]/EC50)nH), where Pomax is the estimated maximum Po for the data set, [X2+] is [Ca2+] or [Cd2+], EC50 is the [Ca2+] or [Cd2+] yielding half-maximal activation and nH is the Hill coefficient. EC50 values reported for each mutant are thus the means of EC50 values determined from individual patches±s.e.m. Electrophysiological results are based on observations from a minimum of five different lipid bilayer patches for each recording condition and includes a total of 324 individual data sets from 67 different lipid bilayer patches.
Crystallization
WT and mutant MthK RCK domains, containing a C-terminal thrombin cleavage site followed by a 6xHis-tag, were expressed in E. coli BL-21(DE3) cells on induction with 0.4 mM IPTG16. Bacteria were collected and resuspended in 20 mM Tris, 250 mM NaCl, pH 7.6 (Buffer B), and lysed by sonication in the presence of phenylmethylsulphonyl fluoride and a protease inhibitor cocktail (Complete EDTA-free, Roche), followed by centrifugation at 30,500 g for 45 min. After adjusting the pH to 7.6, the supernatant was loaded onto a Co2+-charged HiTrap metal affinity column (GE Healthcare), washed with 20 mM imidazole in Buffer B and the channel protein was eluted using 400 mM imidazole in Buffer B. The 6xHis-tag was cleaved immediately after elution by incubating with 2.0 units of thrombin/3.0 mg eluted protein for 2 h at room temperature. The digested protein was then loaded onto a Superdex 200 gel filtration column and eluted with 20 mM Tris, 250 mM NaCl, pH 8.0. The eluted protein solution was concentrated to 6 mg ml−1 using a centrifugal concentrator (10 kDa molecular weight cut-off value) and the concentrated protein was aliquotted and stored in liquid N2 until use. Crystals were formed in 2-μl hanging drops consisting of equal volumes of protein solution (6 mg ml−1 of purified MthK RCK domain, 250 mM NaCl, 20 mM Tris, pH 8.0) and crystallization buffer (6–12% polyethylene glycol 4,000, 1 M ammonium formate, 100 mM sodium acetate, pH 4.8, and 0.2–1 M calcium chloride, as indicated). Calcium chloride concentrations in the crystallization buffers were (in M): WT-C1, 0.2; E212Q, 0.2; D184N, 0.3; and WT-C1C2C3, 1.0. Crystals were mounted in nylon loops and rapidly frozen in liquid N2 without additional cryoprotection, and then stored in liquid N2 until the time of diffraction data collection.
X-ray data collection and analysis
Diffraction data were collected at beamline X6A (WT-C1) or X25 (WT-C1C2C3, D184N and E212Q) at the National Synchrotron Light Source, at 100 K under a nitrogen stream, at a wavelength of 1.1 Å. Data were processed and scaled using HKL2000 or iMosflm, and phasing was done by molecular replacement with Phaser using the unliganded MthK RCK domain structure (PDB accession number 2AEJ) as the search model38,39. The final models for WT-C1 and WT-C1C2C3 each contained one RCK dimer (two peptide chains) per asymmetric unit, and final models for the D184N and E212Q mutants contained three RCK dimers (six peptide chains) per asymmetric unit. Examination of electron density maps and additional model building was performed using COOT, and models were refined by positional, isotropic B-factor and Translation/Libration/Screw refinement in Phenix40,41. To increase the observation:parameter ratio, torsion-angle non-crystallographic symmetry restraints were included in refinement for all but the WT-C1 structure. For the D184N and E212Q structures, which were refined using six NCS groups, bias in the assignment of the Rfree test sets was minimized by the use of thin resolution shells in test set selection, also performed using Phenix. Ramachandran plot analysis for each structure yielded (% in most favoured, allowed and disallowed regions): WT-C1C2C3 (98.2, 1.8, 0.0), WT-C1 (97.3, 2.7, 0.0), D184N (98.7, 1.3, 0.0) and E212Q (98.6, 1.4, 0.0). Omit maps were generated by deleting Ca2+ ions and surrounding side chains, followed by two cycles of simulated annealing refinement, using Phenix. Figures were prepared using PyMOL.
Additional information
Accession codes: Coordinates and structure factors have been deposited in the RCSB protein databank under accession codes 4L73, 4L74, 4L75 and 4L76.
How to cite this article: Smith, F. J. et al. Structural basis of allosteric interactions among Ca2+-binding sites in a K+-channel RCK domain. Nat. Commun. 4:2621 doi: 10.1038/ncomms3621 (2013).
References
Cao, Y. et al. Gating of the TrkH ion channel by its associated RCK protein TrkA. Nature 496, 317–322 (2013).
Kuo, M. M. C., Haynes, W. J., Loukin, S. H., Kung, C. & Saimi, Y. Prokaryotic K+ channels: from crystal structures to diversity. FEMS Microbiol. Rev. 29, 961–985 (2005).
Kroning, N. et al. ATP binding to the KTN/RCK subunit KtrA from the K+-uptake system KtrAB of Vibrio alginolyticus: its role in the formation of the KtrAB complex and its requirement in vivo. J. Biol. Chem. 282, 14018–14027 (2007).
Jiang, Y. et al. Crystal structure and mechanism of a calcium-gated potassium channel. Nature 417, 515–522 (2002).
Wei, A., Solaro, C., Lingle, C. & Salkoff, L. Calcium sensitivity of BK-type KCa channels determined by a separable domain. Neuron 13, 671–681 (1994).
Zhang, X., Zeng, X., Xia, X. M. & Lingle, C. J. pH-regulated Slo3 K+ channels: properties of unitary currents. J. Gen. Physiol. 128, 301–315 (2006).
Parfenova, L. V., Abarca-Heidemann, K., Crane, B. M. & Rothberg, B. S. Molecular architecture and divalent cation activation of TvoK, a prokaryotic potassium channel. J. Biol. Chem. 282, 24302–24309 (2007).
Miranda, P. et al. State-dependent FRET reports calcium- and voltage-dependent gating-ring motions in BK channels. Proc. Natl Acad. Sci. USA 110, 5217–5222 (2013).
Hou, S., Xu, R., Heinemann, S. H. & Hoshi, T. Reciprocal regulation of the Ca2+ and H+ sensitivity in the SLO1 BK channel conferred by the RCK1 domain. Nat. Struct. Mol. Biol. 15, 403–410 (2008).
Pau, V. P., Abarca-Heidemann, K. & Rothberg, B. S. Allosteric mechanism of Ca2+ activation and H+-inhibited gating of the MthK K+ channel. J. Gen. Physiol. 135, 509–526 (2010).
Kong, C. et al. Distinct gating mechanisms revealed by the structures of a multi-ligand gated K(+) channel. eLife 1, e00184 (2012).
Sweet, T. B. & Cox, D. H. Measurements of the BKCa channel's high-affinity Ca2+ binding constants: effects of membrane voltage. J. Gen. Physiol. 132, 491–505 (2008).
Hu, L., Yang, H., Shi, J. & Cui, J. Effects of multiple metal binding sites on calcium and magnesium-dependent activation of BK channels. J. Gen. Physiol. 127, 35–49 (2006).
Jiang, Y. et al. The open pore conformation of potassium channels. Nature 417, 523–526 (2002).
Niu, X., Qian, X. & Magleby, K. L. Linker-gating ring complex as passive spring and Ca(2+)-dependent machine for a voltage- and Ca(2+)-activated potassium channel. Neuron 42, 745–756 (2004).
Pau, V. P. et al. Structure and function of multiple Ca2+-binding sites in a K+ channel regulator of K+ conductance (RCK) domain. Proc. Natl Acad. Sci. USA 108, 17684–17689 (2011).
Ye, S., Li, Y., Chen, L. & Jiang, Y. Crystal structures of a ligand-free MthK gating ring: insights into the ligand gating mechanism of K+ channels. Cell 126, 1161–1173 (2006).
Parfenova, L. V., Crane, B. M. & Rothberg, B. S. Modulation of MthK potassium channel activity at the intracellular entrance to the pore. J. Biol. Chem. 281, 21131–21138 (2006).
Li, Y., Berke, I., Chen, L. & Jiang, Y. Gating and inward rectifying properties of the MthK K+ channel with and without the gating ring. J. Gen. Physiol. 129, 109–120 (2007).
Zadek, B. & Nimigean, C. M. Calcium-dependent gating of MthK, a prokaryotic potassium channel. J. Gen. Physiol. 127, 673–685 (2006).
Smith, F. J., Pau, V. P., Cingolani, G. & Rothberg, B. S. Crystal structure of a Ba(2+)-bound gating ring reveals elementary steps in RCK domain activation. Structure 20, 2038–2047 (2012).
Kuo, M. M., Baker, K. A., Wong, L. & Choe, S. Dynamic oligomeric conversions of the cytoplasmic RCK domains mediate MthK potassium channel activity. Proc. Natl Acad. Sci. USA 104, 2151–2156 (2007).
Dvir, H., Valera, E. & Choe, S. Structure of the MthK RCK in complex with cadmium. J. Struct. Biol. 171, 231–237 (2010).
Schreiber, M. & Salkoff, L. A novel calcium-sensing domain in the BK channel. Biophys. J. 73, 1355–1363 (1997).
Xia, X. M., Zeng, X. & Lingle, C. J. Multiple regulatory sites in large-conductance calcium-activated potassium channels. Nature 418, 880–884 (2002).
Zhang, G. et al. Ion sensing in the RCK1 domain of BK channels. Proc. Natl Acad. Sci. USA 107, 18700–18705 (2010).
Striegel, A. R. et al. Calcium binding by synaptotagmin's C2A domain is an essential element of the electrostatic switch that triggers synchronous synaptic transmission. J. Neurosci. 32, 1253–1260 (2012).
Dong, J., Shi, N., Berke, I., Chen, L. & Jiang, Y. Structures of the MthK RCK domain and the effect of Ca2+ on gating ring stability. J. Biol. Chem. 280, 41716–41724 (2005).
Strynadka, N. C. & James, M. N. Crystal structures of the helix-loop-helix calcium-binding proteins. Annu. Rev. Biochem. 58, 951–998 (1989).
Liriano, M. A. et al. Target binding to S100B reduces dynamic properties and increases Ca(2+)-binding affinity for wild type and EF-hand mutant proteins. J. Mol. Biol. 423, 365–385 (2012).
Carter, P. J., Winter, G., Wilkinson, A. J. & Fersht, A. R. The use of double mutants to detect structural changes in the active site of the tyrosyl-tRNA synthetase (Bacillus stearothermophilus). Cell 38, 835–840 (1984).
Cox, D. H. The BKCa channel's Ca2+-binding sites, multiple sites, multiple ions. J. Gen. Physiol. 125, 253–255 (2005).
Zeng, X. H., Xia, X. M. & Lingle, C. J. Divalent cation sensitivity of BK channel activation supports the existence of three distinct binding sites. J. Gen. Physiol. 125, 273–286 (2005).
Qian, X., Niu, X. & Magleby, K. L. Intra- and intersubunit cooperativity in activation of BK channels by Ca2+. J. Gen. Physiol. 128, 389–404 (2006).
Cui, J., Yang, H. & Lee, U. S. Molecular mechanisms of BK channel activation. Cell. Mol. Life Sci. 66, 852–875 (2009).
Heginbotham, L., Kolmakova-Partensky, L. & Miller, C. Functional reconstitution of a prokaryotic K+ channel. J. Gen. Physiol. 111, 741–749 (1998).
Thomson, A. S. & Rothberg, B. S. Voltage-dependent inactivation gating at the selectivity filter of the MthK K+ channel. J. Gen. Physiol. 136, 569–579 (2010).
Otwinowski, Z. & Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 (1997).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, (Pt 4): 658–674 (2007).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta. Crystallogr. D Biol. Crystallogr. 60, (Pt 1, 12): 2126–2132 (2004).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta. Crystallogr. D Biol. Crystallogr. 66, (Pt 2): 213–221 (2010).
Acknowledgements
We thank Chris Lingle (Washington University in St Louis) and Andrew Thomson (Temple University) for helpful discussions and comments on the manuscript, and Vivian Stojanoff, Annie Héroux and the staff of NSLS beamlines X6A and X25 for assistance. This work was supported by the National Science Foundation under Grant No. MCB-1243803 to B.S.R.
Author information
Authors and Affiliations
Contributions
F.J.S. and V.P.T.P. performed electrophysiological experiments and data analysis. F.J.S. performed biochemical and all crystallization experiments. G.C. performed crystallographic data collection and analysis for the WT-C1 crystal, and F.J.S. and B.S.R. performed all other crystallographic data collection and analysis, and refinement of all structures. F.J.S. and B.S.R. wrote the manuscript with input from the other authors. B.S.R. supervised the project.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Smith, F., Pau, V., Cingolani, G. et al. Structural basis of allosteric interactions among Ca2+-binding sites in a K+ channel RCK domain. Nat Commun 4, 2621 (2013). https://doi.org/10.1038/ncomms3621
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms3621
This article is cited by
-
Calcium-gated potassium channel blockade via membrane-facing fenestrations
Nature Chemical Biology (2024)
-
Structure of potassium channels
Cellular and Molecular Life Sciences (2015)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
