
Abstract
Located off the northwestern coast of the European mainland, Britain and Ireland were among the last regions of Europe to be colonized by modern humans after the last glacial maximum. Further, the geographical location of Britain, and in particular of Ireland, is such that the impact of historical migration has been minimal. Genetic diversity studies applying the Y chromosome and mitochondrial systems have indicated reduced diversity and an increased population structure across Britain and Ireland relative to the European mainland. Such characteristics would have implications for genetic mapping studies of complex disease. We set out to further our understanding of the genetic architecture of the region from the perspective of (i) population structure, (ii) linkage disequilibrium (LD), (iii) homozygosity and (iv) haplotype diversity (HD). Analysis was conducted on 3654 individuals from Ireland, Britain (with regional sampling in Scotland), Bulgaria, Portugal, Sweden and the Utah HapMap collection. Our results indicate a subtle but clear genetic structure across Britain and Ireland, although levels of structure were reduced in comparison with average cross-European structure. We observed slightly elevated levels of LD and homozygosity in the Irish population compared with neighbouring European populations. We also report on a cline of HD across Europe with greatest levels in southern populations and lowest levels in Ireland and Scotland. These results are consistent with our understanding of the population history of Europe and promote Ireland and Scotland as relatively homogenous resources for genetic mapping of rare variants.
Similar content being viewed by others

Introduction
Over the past 5 years, rapid technological developments in high-throughput genotyping, and more recently sequencing, have heralded a true era of population genomics. The high-resolution data sets that suddenly became available accelerated the development of analytical approaches for exploring questions surrounding signatures of population history (eg, linkage disequilibrium (LD), population structure, admixture), how selection has shaped the architecture of our genome, and for crucial identification of genetic factors predicting the development and treatment of common human diseases.
It is on the question of identifying the common genetic variants influencing risk of common diseases such as type 2 diabetes and cancer that most resources have been focused. High-density single-nucleotide polymorphism (SNP) data sets have rapidly accelerated and refined genetic mapping techniques and reducing costs have meant that strategies, such as association, linkage, admixture and homozygosity mapping, have been applied with notable success. Before the genome-wide association study era, the community of human geneticists could agree on only a handful of replicated genetic risk factors for complex disease, whereas today there is agreement on over 1000 and the number continues to rise.1 However, despite this progress, much work remains to be carried out towards understanding the genetic architecture involved. For example, despite very large and well-powered studies, much of the genetic contribution towards disease and trait prediction remains to be characterized; this has been dubbed as the ‘missing heritability’.2 It is also clear that the variants identified through genome-wide association studies are in most cases not the causal variants, and considerable fine mapping of known associations will be required before an accurate catalogue of even the most common variation influencing human health and disease can be collated.
An improved understanding of the population genetic structure can refine and empower the application of genetic mapping. For example, differences in patterns of LD across populations can compromise the expected performance of genome-wide association study experiments; cryptic stratification (even when controlled for) will further compromise power, whereas incorporating population admixture into the study design can lead to significantly increased power.3, 4
In this paper, we seek to further our knowledge of the population structure of Britain and Ireland. Located off the western seaboard of the European mainland, these islands are of particular interest as they represent one of the last regions of Europe to be populated continuously. Further, at the northwestern extreme of the continent and isolated by sea, these islands are geographically remote. This relative isolation and recent population history may have resulted in particular genomic traits that are advantageous for genetic mapping (eg, extended LD, lower allelic diversity, increased homozygosity and so on). Y chromosome analysis has revealed a rather low diversity in the region compared with other parts of Europe, with signals of continuity from the earliest times and relatively little subsequent input from settlers in the meantime, with major contributions limited to particular areas of Britain, including the north and west of Scotland and the eastern seaboard of Britain.5, 6, 7 Moreover, the recent discovery of Y chromosome variants with very circumscribed distributions across the isles (JF Wilson, unpublished) indicates considerable isolation among the populations inhabiting Ireland and those that are now England and Scotland. The most striking pattern in Scotland and England is a strong east-to-west differentiation, a palimpsest resulting no doubt from multiple immigrations from the east.8 Mitochondrial DNA analysis also reveals lower than average diversity in Ireland, but not Britain.9 Using a data set of 4424 SNPs, we previously confirmed the close genetic relationship between Irish and neighbouring European populations and illustrated the suitability of the HapMap Utah Centre d'Etude du Polymorphisme Humain (CEPH) data set for the design of LD-based genetic mapping studies in the Irish population.10
In this study, using whole-genome association data from the Irish, British and mainland European populations, we seek to provide a detailed description of the patterning of common human genetic variation with a view to informing future large-scale genetic mapping studies. We focus on four key areas—population genetic structure, LD, homozygosity and genetic diversity. Through principal components and Bayesian approaches, we explore the structuring of genetic variation within and among British, Irish and European populations. We characterize the patterning of LD across the different samples and compare it with the HapMap European-heritage population with the aim of identifying and understanding subtle among-population differences in LD that might influence the efficiency of genetic mapping. We investigated the nature and extent of runs of homozygosity (ROH) in each population. Finally, we calculated summary statistics of haplotype diversity (HD) with a view to commenting on disease allele diversity and the corresponding influence on genetic mapping.
Materials and methods
Samples
We used genotype data from a total of 3367 individuals recruited from seven different European or European descent populations. They were Ireland/Dublin (N=866), Scotland/Aberdeen (N=702), Bulgaria (N=611), Portugal (N=216), Sweden (N=400), South/Southeast England (N=512) and Utah (N=60). The Irish/Dublin, Scottish/Aberdeen, Bulgarian, Portuguese and Swedish populations were recruited as healthy controls, as part of the International Schizophrenia Consortium's (ISC) genetic mapping effort. Further details on the ISC population's samples are available elsewhere.11 Genotypes of the 512 British samples were accessed from the Wellcome Trust Case Control Consortium (WTCCC) 1958 Birth Control Data set. The Utah European ancestry population (CEU) was from the HapMap project, phase 2. To provide structured coverage of Great Britain, we subdivided the 1958/WTCCC samples (on the basis of postcode information) as either ‘southern’ (approximately Dorset, Hampshire, Oxfordshire, Berkshire) or ‘southeastern’ (approximately Kent, Surry, East and West Sussex), as defined by the WTCCC.12 The ‘southern’ and ‘southeastern’ populations were selected to complement the more ‘northern’ Scottish population.
The ISC samples were all genotyped on the Affymetrix (Santa Clara, CA, USA) 5.0 and 6.0 platforms (739 995 SNPs). We cleaned this data set by excluding individuals with >10% missing genotypes and SNPs with minor allele frequencies <1%. To allow merging with the HapMap and WTCCC (Affy 5.0) data sets, we reduced the ISC data set to the core set of 344 901 SNPs common to all HapMap, WTCCC and ISC populations investigated in this study.
Population structure
Principal components analysis (PCA) was carried out using the Eigensoft package (v2.0).4, 13 We applied the software using parameters of five iterations of outlier removal, and calculated 20 eigenvectors or principal components in each analysis. We visualized these data using kernel density plots created by the Ecological Data Analysis (ADE4) package in R.14 In addition to PCA, we assessed the population structure using the program FRAPPE,15 predicting individual ancestry for a range of assumed numbers of distinct populations (K). PCA and FRAPPE analyses were carried out on 60 random individuals from each population (matching the number of HapMap CEU founders used and for computational efficiency). Data were visualized using the program diSTRUCT.16
LD
Pairwise D′ and r2 values were calculated within populations using PLINK.17 To account for the effects of unequal sample sizes, we selected 60 random individuals to represent each population. To compare the range and extent of LD across our populations, we calculated mean LD for each pair using 3850 SNPs along chromosome 22 within 1000 bp bins (ie, bin 1=0–1000, bin 2=1001–2000 and so on). Confirmatory analysis of LD patterns observed on chromosome 22 was conducted using genotype data from chromosome 16. We plotted LD against physical distance for SNP pairs within 0–70 kb.18
LD unit (LDU) maps are a useful way of summarizing patterns of LD in populations, where LDU represents the product of the physical distance between SNPs and a parameter that reflects the decline in the probability of association between markers according to physical distance.19 We calculated LDU maps for each population using chromosome 22 genotypes and previously described methodology.20 Consistent with the literature, we defined LD ‘holes’ as regions with a gap greater than 2.5 LDU between adjacent SNPs.19, 21
ROH
ROH were identified using PLINK v1.06.17 We applied the following parameters for calculating ROH: 5 MB window size; a minimum of 50 SNPs per window; and allowing one heterozygous and five missing calls per window. In addition, we used a minimum length cutoff of 1 Mb and a minimum number of 100 contiguous homozygous SNPs. We excluded regions in which the mean tract density exceeded 50 kb per SNP and used a maximum gap between two consecutive homozygous SNPs of 1 Mb.21, 22, 23 ROH were identified separately for each population. We assessed three features of ROH—(i) number of homozygous segments (average and range, calculated across individuals within a population), (ii) summed segment length (average and range, calculated across individuals within a population) and (iii) FROH, a genomic measure of individual autozygosity, defined as the proportion of the autosomal genome in ROH above a specified length threshold22 (we use FROH1 to define the proportion of the genome in runs 1 Mb or greater in length, and FROH5 to define proportions in runs of 5 Mb or greater in length).
HD
We estimated HD within each population (N=60, same sets as for LD analysis) by the average number of haplotypes observed in nonoverlapping 0.5 cM windows spanning the autosomal genome, as previously described.24 Haplotypes were phased using Beagle v3.0.4.25 The genome was divided into 0.5 cM windows, in which the first SNP on each chromosome defined the start of window 1 and the next window started at the position at which the preceeding window ended. Haplotypes across each of our populations were counted from random selections of either 10 SNPs (when between 10 and 25 SNPs were found within a bin) or 25 SNPs (when more than 25 SNPs were found). Windows containing fewer than 10 SNPs were discarded. We then summarized HD for each of the populations separately and interpreted our findings in the context of previously reported results from additional European populations.24
Results
Analysis of population structure
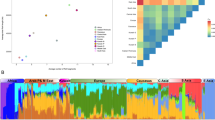
Eigensoft PCA analysis across all seven of our European and European-ancestry populations broadly identified four sub-groups consisting of (i) Bulgarian, (ii) Portuguese, (iii) Swedish and (iv) Irish/British/Utah populations (see Figure 1a, Supplementary Figure S1). The first two principal components (PCs) separate out northern from southern, and western from eastern European ancestry, respectively. The Europe-wide PCA analysis positions the Scottish population (Aberdeen) intermediate between the Irish and English populations. We further explored this observation by restricting our PC analysis to residents of Ireland, Scotland (Aberdeen) and south/southeast England (Figure 1b, Supplementary Figure S1). This analysis confirms the observation that the Scottish population is intermediate between the Irish and English cohorts on the first principal component (this time dividing west from east). Although more subtle, the Scottish cohort is also shifted slightly from the other two on PC2.

PCA results for British, Irish and European populations. (a) All populations, (b) Irish and British populations only. ABER, Scotland/Aberdeen; DUBL, Ireland/Dublin; ENGL_S_SE, England South and Southeast combined; PORT, Portugal; BULG, Bulgaria; SWED, Sweden; CEU, Utah HapMap European Americans. A similar PCA plot has been generated from independent analysis of the ISC data, which has a different focus, paper submitted.
We next explored genetic differentiation using the model-based Bayesian clustering approach incorporated in the program FRAPPE.15 As expected, results overlapped closely with the PC analysis. Restricting analysis to 60 individuals from each of the eight populations, we observed the effect of increasing k (the number of specified sub-populations to be inferred). The first component of our PC analysis (north/south division) roughly corresponds with the k=2 FRAPPE analysis, with Bulgaria and Portugal separating from the other populations. At k=3, we see Sweden separating out from the other northern European populations. At k=4, we can resolve Bulgarian, Portuguese and Swedish individuals from Britain and Ireland. Increasing to k=5 provides little additional resolution, although there seems to be a continuum of variation from the Irish population through to Scottish to English and CEU (see Supplementary Figure S2).
Interpopulation fixation index-statistics (FST) revealed subtle but significant differences between populations, supporting PCA and FRAPPE results (see Table 1). As expected, the largest difference (FST=0.5%) was between Sweden and Portugal (who seem distant on both PC1 and PC2, and are identified in the k=2 FRAPPE analysis), followed by Bulgaria and Ireland (who again differ across both PC1 and PC2). The British and Irish populations show very little differentiation (FST∼0.02–0.06%), with the Scottish population seeming to be closer to the southern English than to the Irish.
Patterning of LD
To explore the nature and extent of LD, we calculated within-population r2 and D′ values and compared these across populations. The breakdown and patterning of LD with distance is virtually indistinguishable among the Irish, Scottish, southern English, Swedish and HapMap CEU populations (Supplementary Figure S3). The similarity of LD patterning across populations is not chromosome specific and is repeated when calculated using genotype data from chromosome 16.
We next explored differences in cross-population LD patterning using LDU maps. These maps are generated from the product of physical distance between flanking SNPs and a parameter describing the decline in association with distance computed for each interval. ‘Shorter’ maps represent populations with high levels of LD and the overall map length correlates with (effective) time to a population bottleneck. We included an LDU analysis, as it may be more sensitive to LD patterning than r2 and D′.19 Indeed, although LD patterning across all the European descent populations did not differ strongly in the overall patterning of r2 and D′, we observed subtle but clear differences using LDU maps. Figure 2 illustrates that the Swedish and Irish populations have shorter LDU maps, indicative of more extensive LD across chromosome 22. This difference in LD patterns is also reflected in the estimated number of LD holes in each population. The Swedish population had the lowest overall number (64), followed by Ireland (88), HapMap CEU (97), southern England (105) and Scotland (109) (see Table 2, Supplementary Figure S4).

LDU map for Irish, British and CEU populations. See Figure 1 legend for population identifiers.
ROH
The observation of a subtle degree of isolation for the Irish group at the LD level is also in agreement with the results of our ROH analysis. Again we see very similar patterning across populations, but our results suggest that the Irish population has the largest proportion of the genome in ROH (as measured by FROH1), relative to the British and HapMap CEU populations examined here (Figure 3). Overall, the Irish and Swedish populations seem slightly different from the others in the context of ROH. Both the Irish and Swedish populations showed, on an average, a greater number of ROH, an increased maximum ROH length, as well as an increased proportion of the genome in homozygous runs, compared with that of the Scottish, southern English and Utah populations. Similarly, the mean level of individual autozygosity per population as measured by FROH22 was highest for the Irish group (Figure 4). Together, these results suggest slightly increased autozygosity in the Irish cohort compared with the British and Swedish cohorts.

FROH1 patterning in Irish, British and Swedish populations. Box plots represent (a) the number and (b) the summed size of segments of the autosomal genome that exists in ROH of 1 Mb or greater in length (ie, FROH1). The bars represent mean and confidence intervals, as per a standard box plot (box indicating the 25th–75th percentile of the FROH1 distribution, line within box representing the median and ends of the whiskers representing the 5th–95th percentiles). Outliers are represented by diamonds.

Mean FROH1 and FROH5 patterning in Irish, British and Swedish populations. See Figure 1 legend for population identifiers. Y-axis indicates the average proportion of the autosomal genome covered by FROH1 or FROH5 (see Materials and Methods for definition of FROH).
Autozygosity is generated by increased levels of kinship, which in turn reflects the population history of Ireland. Although relatively undisturbed by secondary migrations,5, 26 the population of Ireland has undergone expansions and contractions at numerous points in recent history (eg, two major famines since 1600, disease epidemics, expansion in the first half of the 19th century). Aside from these features, the increased autozygosity may also reflect legacies of Gaelic family structures and comparatively low levels of migration that are in part due to a lack of industrial revolution in Ireland.
To test a hypothesis of increased autozygosity due to features of relatively recent population history, we examined the patterning of homozygosity looking for signals of parental relatedness over the last four or five generations. Previous work has illustrated that parental relatedness arising within four to six generations predominantly affects ROH over 5 Mb in length.22 We therefore compared this statistic across populations. Results show that the Irish and Swedish populations have around 10 times as much of their genomes in ROH over 5 Mb in length than the southern English, and 1.5–3 times as much as Scotland and Utah (Figure 4).
HD
Our final set of analyses explored the degree of HD (HD) across populations. HD represents a method for quantifying genetic diversity that integrates LD. The nature and patterning of HD can, in theory, reflect the underlying disease-causing genetic variation and provide insight into human demographic history. We estimated HD across our populations and placed our results in the context of previous estimates for European and world populations, calculated using the same technique.24 Diversity across Britain and Ireland is reduced in comparison with mainland European populations, with Scotland and Ireland having lower levels than southern England. The cline in HD across Europe is seen to extend all the way to the Atlantic, where the lowest national genetic diversities in Europe are apparent (Figure 5). These results are in close agreement with an earlier survey of HD across Europe24 and are consistent with a serial founder effect, a scenario in which population expansion involves successive migration of a small fraction of individuals out of the previous location, starting from a single origin in sub-Saharan Africa.27

HD estimates across Europe. Values represent HD in the order of H10, H25 (see Materials and Methods for calculation of H10 and H25). *indicates populations for which H10/H25 values have been taken directly from the literature.24
Discussion
Our analysis of population structure through both PCA and Bayesian approaches reiterates previously reported patterns of genetic structure across Europe. PC1 clearly differentiates northern from southern Europe, whereas PC2 differentiates eastern from western Europe.28, 29, 30 What was perhaps less well described was the subtle structuring of variation across Britain and Ireland. Through PCA we were able to resolve the Irish population from both the Scottish and English populations, consistent with previous observations based on a similarly sized data set.29 Further, we observed subtle differentiation between Scotland and England, as previously illustrated in WTCCC data.12, 31 A previous report of the European population structure that was focused primarily on mainland Europe reported interpopulation FST values of 0.003.28 It is interesting to note that across Britain and Ireland, FST values were an order of magnitude lower (average English/Irish/Scottish value=0.0005). Although intermediate between Irish and English populations by PCA, our Scottish population seems genetically more similar to the English than to the Irish population using FST. This result is in keeping with the geographical proximity between Scotland and England and the sharing, therefore, of more historical and prehistorical influences than with Ireland. The degree of sharing between Scotland and Ireland or England is probably structured according to geography. It would be interesting, for example, to quantify genetic sharing between western Scottish regions (eg, Argyll or Galloway) and Ireland or, conversely, between the Border region and England. Evidence for genetic structure within Scotland exists from Y-chromosome analysis, which reveals both a shared ancestry between eastern Scottish and eastern English samples, such as ours, and a similarity between Scotland and Ireland to the exclusion of England (JF Wilson, unpublished data).
The degree of differentiation observed in this study is conservative, given that samples were not collected on the basis of ancestry, but rather on the basis of residence only. Given the massively increased level of population mobility in the last century, one would expect increased differentiation if sampling were restricted to individuals with all four grandparents from particular regions of interest.
The Utah population is known to have a majority English ancestry. Our results are consistent with this; the HapMap CEU and southern English populations being virtually indistinguishable using both FST and PCA.
In agreement with the population structure results, our comparisons of LD illustrated close parallels between the populations tested in this study. At the level of D′ and r2, the populations seemed indistinguishable, consistent with our own and other authors’ previous results.10, 32 However, it is interesting to note that using the LDU parameter, we observed increased levels of LD and reduced numbers of ‘LD holes’ in Irish and Swedish populations (see Figure 3, Table 2). However, it should be stressed that the increase in LD is marginal. It is well known that population isolates show significantly increased levels of LD. Classic isolates typically generate LDU maps (of chromosome 22) with lengths in the region of 400–700 units.19, 21 Although the corresponding LDU map length of Ireland (858 units) and Sweden (818 units) is shorter than that of Scotland, southern England and Utah CEPH (894–903 units), it is still significantly longer than that of a typical isolate. In this sense, the concept of the general Irish population as anything approaching a population isolate can be dismissed. However, reflecting the situation across Europe, populations showing characteristics of a genetic isolate probably exist within the rural communities of Ireland, or on islands off the mainland. Known European examples of genetic isolates located close to cosmopolitan populations include the town of Rucphen in the Netherlands and the Orkney Islands off Scotland. Focusing research on rural Irish communities would shed further light on this question.
Analysis of ROH is a powerful method to gauge the extent of ancient kinship and recent parental relationship within a population. This is because ROH arise from shared parental ancestry in an individual's pedigree. The offspring of cousins have very long ROH, commonly over 10 Mb, whereas at the other end of the spectrum, almost all Europeans have ROH of ∼2 Mb in length, reflecting shared ancestry from hundreds to thousands of years ago. By focussing on ROH of different lengths, it is therefore possible to infer aspects of demographic history at different time depths in the past.22 We used FROH measures to compare and contrast patterning across populations. These measures are genomic equivalents of the pedigree inbreeding coefficient, but do not suffer from problems of pedigree reconstruction. By varying the lengths of ROH that are counted, they may be tuned to assess parental kinship at different points in the past. We used two different measures, FROH1, which includes all ROH over 1 Mb and hence includes information on recent and background parental relatedness, and FROH5, which sums ROH over 5 Mb in length, more typical of a parental relationship in the last four to six generations.22 Our FROH1 results indicate slightly elevated levels in the Irish and Swedish populations (compared with southern England, Scotland and HapMap CEU) of both the overall number of ROH and the proportion of genome in ROH (see Figure 3). This pattern was exaggerated when we restricted analysis to ROH greater than 5 Mb in length (ie, FROH5, see Figure 4), indicating increased levels of parental relatedness in the last six generations in the Irish and Swedish populations compared with other populations tested in this study. When we remove individuals with ROH over 5 Mb from the FROH1 analysis (Supplementary Figure S5), Ireland remains as the population with the most homozygous runs and the longest sum length of homozygosity. This provides further evidence that the elevated proportion of shorter ROH, and hence the number of ancient pedigree loops in Ireland, is indeed real and not driven by a limited number of offspring of cousins.
Famine and mass emigration may have driven the increased levels of autozygosity in the Irish population. However, we consider it likely that the increased levels we have observed are at least partially attributable to the genetic remnants of ancient Gaelic patrilineal dynasties,33, 34, 35 in combination with the traditionally agricultural nature of Irish society. Ireland was not affected by industrial revolution to the same extent as Britain. Industrial revolution has been associated with mass migration from rural to urban communities and an expansion of effective population size. The absence of such a pattern in Ireland would have resulted in an extended adherence to primogeniture-style inheritance of land, with frequently only one adult sibling from each family being enabled to marry and reproduce by accession to farm ownership. Such patterns would have restricted growth in effective population size. However, a potential confounder in this study is the sampling scheme: at least in areas with little mobility, recruiting primarily from rural as opposed to urban areas could increase levels of autozygosity, as could sampling a group of people who were born earlier.36
Similar to Ireland, Sweden has also traditionally been an agricultural society. Although the industrial revolution drove urbanization in Sweden towards the end of the 19th century, the more recent time to population bottleneck (as indicated by the LDU map) and the resulting rapid expansion during the 20th century might be contributing to the lack of diversity we observe in our data. Further, work taking these variables into account will be necessary to fully understand the causes underlying the patterns we see.
The results of our HD analysis are consistent with expectations from our understanding of European population history. They recapitulate the famous cline in allele frequencies first noted by Ammerman and Cavalli-Sforza37 and associated by them with the spread of Neolithic farmers across the continent. Data from both Y chromosomes38 and mtDNA39 reveal the same patterns. Simulations have since shown that a cline in both allele frequencies and genetic diversity from southeast to northwest Europe would also be expected from the original dispersal of hunter–gatherers into Europe.40 It is likely that both processes contributed, along with postglacial expansions from southern refugia, for example, in Iberia, where diversity is clearly higher in our data set. Similar geographical patterns are seen using SNP heterozygosity.28 Our analysis is the first to reveal that, at the autosomal level, this diversity gradient extends across Britain and Ireland, reaching a low point in Scotland and Ireland, at the edge of the Atlantic.
In summary, our results illustrate a subtle genetic structure across Britain and Ireland in the context of the comparatively homogenous nature of the European genetic pool. We have observed slightly elevated levels of LD and genome-wide homozygosity in Ireland and Sweden compared with neighbouring British and European populations, although these levels do not approach those of traditional population isolates. Similarly, we have illustrated a decrease in HD in Britain and Ireland, more so in Scotland and Ireland than in England. All these characteristics can be advantageous for genetic mapping. A reduced structure moderates the issue of cryptic stratification, although appropriate corrective steps should always be taken. Elevated LD and fewer LD holes will enhance the efficiency and power of genome-wide association study platforms. Reduced HD should reflect the disease allele architecture. Increased ROH will improve power for the identification of recessive effects. Aside from promoting Ireland and Scotland as resources for genetic mapping, the elevated levels of kinship illustrated in Ireland, and potentially in Scotland, would in theory make these particularly amenable populations to apply long-range phasing and haplotype imputation methods for rare variation.41
References
Hindorff LA, Junkins HA, Mehta JP, Manolio TA : A Catalog of Published Genome-Wide Association Studies; Available at: www.genome.gov/gwastudies . Accessed 07/Dec/09.
Manolio TA, Collins FS, Cox NJ et al: Finding the missing heritability of complex diseases. Nature 2009; 461: 747–753.
Tian C, Gregersen PK, Seldin MF : Accounting for ancestry: population substructure and genome-wide association studies. Hum Mol Genet 2008; 17: R143–R150.
Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D : Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 2006; 38: 904–909.
Hill EW, Jobling MA, Bradley DG : Y-chromosome variation and Irish origins. Nature 2000; 404: 351–352.
Capelli C, Redhead N, Abernethy JK et al: A Y chromosome census of the British Isles. Curr Biol 2003; 13: 979–984.
Wilson JF, Weiss DA, Richards M, Thomas MG, Bradman N, Goldstein DB : Genetic evidence for different male and female roles during cultural transitions in the British Isles. Proc Natl Acad Sci USA 2001; 98: 5078–5083.
Richards M, Capelli C, Wilson JF : Encyclopaedia of Life Sciences. John Wiley & Sons: Chichester, 2008.
Helgason A, Hrafnkelsson B, Gulcher JR, Ward R, Stefansson K : A populationwide coalescent analysis of Icelandic matrilineal and patrilineal genealogies: evidence for a faster evolutionary rate of mtDNA lineages than Y chromosomes. Am J Hum Genet 2003; 72: 1370–1388.
O’Dushlaine CT, Dolan C, Weale ME et al: An assessment of the Irish population for large-scale genetic mapping studies involving epilepsy and other complex diseases. Eur J Hum Genet 2008; 16: 176–183.
ISC: Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature 2008; 455: 237–241.
WTCCC: Genome-wide association study of 14 000 cases of seven common diseases and 3000 shared controls. Nature 2007; 447: 661–678.
Patterson N, Price AL, Reich D : Population structure and eigenanalysis. PLoS Genet 2006; 2: e190.
Dray S, Dufour AB : The ade4 package: implementing the duality diagram for ecologists. J Stat Soft 2007; 22: 1–20.
Tang H, Peng J, Wang P, Risch NJ : Estimation of individual admixture: analytical and study design considerations. Genet Epidemiol 2005; 28: 289–301.
Rosenberg NA, Pritchard JK, Weber JL et al: Genetic structure of human populations. Science 2002; 298: 2381–2385.
Purcell S, Neale B, Todd-Brown K et al: PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81: 559–575.
Jakobsson M, Scholz SW, Scheet P et al: Genotype, haplotype and copy-number variation in worldwide human populations. Nature 2008; 451: 998–1003.
Service S, DeYoung J, Karayiorgou M et al: Magnitude and distribution of linkage disequilibrium in population isolates and implications for genome-wide association studies. Nat Genet 2006; 38: 556–560.
Maniatis N, Collins A, Xu CF et al: The first linkage disequilibrium (LD) maps: delineation of hot and cold blocks by diplotype analysis. Proc Natl Acad Sci USA 2002; 99: 2228–2233.
Jakkula E, Rehnstrom K, Varilo T et al: The genome-wide patterns of variation expose significant substructure in a founder population. Am J Hum Genet 2008; 83: 787–794.
McQuillan R, Leutenegger AL, Abdel-Rahman R et al: Runs of homozygosity in European populations. Am J Hum Genet 2008; 83: 359–372.
Lencz T, Lambert C, DeRosse P et al: Runs of homozygosity reveal highly penetrant recessive loci in schizophrenia. Proc Natl Acad Sci USA 2007; 104: 19942–19947.
Auton A, Bryc K, Boyko AR et al: Global distribution of genomic diversity underscores rich complex history of continental human populations. Genome Res 2009; 19: 795–803.
Browning SR, Browning BL : Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet 2007; 81: 1084–1097.
McEvoy B, Richards M, Forster P, Bradley DG : The Longue Duree of genetic ancestry: multiple genetic marker systems and Celtic origins on the Atlantic facade of Europe. Am J Hum Genet 2004; 75: 693–702.
Li JZ, Absher DM, Tang H et al: Worldwide human relationships inferred from genome-wide patterns of variation. Science 2008; 319: 1100–1104.
Lao O, Lu TT, Nothnagel M et al: Correlation between genetic and geographic structure in Europe. Curr Biol 2008; 18: 1241–1248.
McEvoy BP, Montgomery GW, McRae AF et al: Geographical structure and differential natural selection amongst North European populations. Genome Res 2009.
Novembre J, Johnson T, Bryc K et al: Genes mirror geography within Europe. Nature 2008; 456: 98–101.
Burton PR, Clayton DG, Cardon LR et al: Association scan of 14 500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat Genet 2007; 39: 1329–1337.
Cronin S, Berger S, Ding J et al: A genome-wide association study of sporadic ALS in a homogenous Irish population. Hum Mol Genet 2008; 17: 768–774.
McEvoy B, Simms K, Bradley DG : Genetic investigation of the patrilineal kinship structure of early medieval Ireland. Am J Phys Anthropol 2008; 136: 415–422.
Moore LT, McEvoy B, Cape E, Simms K, Bradley DG : A Y-chromosome signature of hegemony in Gaelic Ireland. Am J Hum Genet 2006; 78: 334–338.
King TE, Jobling MA : Founders, drift, and infidelity: the relationship between Y chromosome diversity and patrilineal surnames. Mol Biol Evol 2009; 26: 1093–1102.
Nalls MA, Wilson JG, Patterson NJ et al: Admixture mapping of white cell count: genetic locus responsible for lower white blood cell count in the Health ABC and Jackson Heart studies. Am J Hum Genet 2008; 82: 81–87.
Ammerman A, Cavalli-Sforza LL : The Neolithic Transition and the Genetics of Populations in Europe. Princeton University Press: Princeton, New Jersey, USA, 1984.
Semino O, Passarino G, Oefner PJ et al: The genetic legacy of Paleolithic Homo sapiens sapiens in extant Europeans: a Y chromosome perspective. Science 2000; 290: 1155–1159.
Richards M, Macaulay V, Torroni A, Bandelt HJ : In search of geographical patterns in European mitochondrial DNA. Am J Hum Genet 2002; 71: 1168–1174.
Currat M, Excoffier L : The effect of the Neolithic expansion on European molecular diversity. Proc Biol Sci 2005; 272: 679–688.
Kong A, Masson G, Frigge ML et al: Detection of sharing by descent, long-range phasing and haplotype imputation. Nat Genet 2008.
Acknowledgements
We acknowledge the use of genotype data from the British 1958 Birth Cohort DNA collection, funded by the Medical Research Council Grant G0000934 and the Wellcome Trust Grant 068545/Z/02. CTO’D was funded by a post-doctoral fellowship for the Irish Research Council for Science Engineering and Technology, JW by the Royal Society and AC from Science Foundation Ireland. We thank three anonymous reviewers for their constructive and helpful comments.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Rights and permissions
About this article
Cite this article
O'Dushlaine, C., Morris, D., Moskvina, V. et al. Population structure and genome-wide patterns of variation in Ireland and Britain. Eur J Hum Genet 18, 1248–1254 (2010). https://doi.org/10.1038/ejhg.2010.87
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2010.87
Keywords
This article is cited by
-
Weighted likelihood inference of genomic autozygosity patterns in dense genotype data
BMC Genomics (2017)
-
The Irish DNA Atlas: Revealing Fine-Scale Population Structure and History within Ireland
Scientific Reports (2017)
-
Iron Age and Anglo-Saxon genomes from East England reveal British migration history
Nature Communications (2016)
-
Genetic structure of the Newfoundland and Labrador population: founder effects modulate variability
European Journal of Human Genetics (2016)
-
Recent genomic heritage in Scotland
BMC Genomics (2015)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}