


I. INTRODUCTION
Cerium oxide is widely investigated for its capability of forming solid solutions in the whole composition range with oxides of rare earths, transition metals (Minervini et al., Reference Minervini, Zacate and Grimes1999; Foschini et al., Reference Foschini, Souza, Paulin Filho and Varela2001), and group III cations (Park et al., Reference Park, Sohn and Butt2000; Gregori et al., Reference Gregori, Rahmati, Sigle, van Aken and Maier2011). Particularly sought for is doping effected by substituting Ce4+ with trivalent cations from sesquioxides, in order to introduce oxygen vacancies according to the equilibrium Eq. (1):
 $$\displaystyle{1 \over 2}\, {M_2}{O_3}\buildrel {\hbox{Ce}\, \, {\hbox{O}_2}} \over \longrightarrow M_{{\rm Ce}}^{\prime}+\displaystyle{1 \over 2}V_{\ddot{o}} -+\displaystyle{3 \over 2}{O_o}$$
$$\displaystyle{1 \over 2}\, {M_2}{O_3}\buildrel {\hbox{Ce}\, \, {\hbox{O}_2}} \over \longrightarrow M_{{\rm Ce}}^{\prime}+\displaystyle{1 \over 2}V_{\ddot{o}} -+\displaystyle{3 \over 2}{O_o}$$
Oxide ion vacancies are wanted for those applications requiring ionic conductivity. Charge transport and, in turn, ionic conductivity have an inverse dependence on the activation energy needed by the vacancies for migrating in the lattice. To this point, the crystal environment around the vacancies is of primary importance. In case of extended clustering of defects, oxygen vacancies would be trapped nearby the dopant cations in an energetically favourable environment (Goodenough, Reference Goodenough2003). High activation energy barrier would ensue, resulting in low ionic conductivity. Conversely, if vacancies were not associated but scattered in the CeO2 structure, instead, they would be relatively free to migrate, thus, promoting ionic conductivity.
There is no agreement in the literature on how and even if the defects in doped ceria are aggregated in space. Conclusions are mainly biased by the techniques employed to investigate the defect structure. Bulk probes such as EXAFS and solid-state NMR (Yamazaki et al., Reference Yamazaki, Matsui, Ohashi and Arita2000; Kim and Stebbins, Reference Kim and Stebbins2007) suggest the formation of trimers
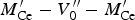 $M_{{\rm Ce}}^{\prime} - V_0^{{\prime}{\prime}} - M_{{\rm Ce}}^{\prime}$
; however, such techniques only investigate the first few coordination shells of an atom. At odds with that, electron microscopy probes such as transmission electron microscopy (TEM), selected area electron diffraction (SAED), and electron energy loss spectroscopy (EELS) (Ou et al., Reference Ou, Mori, Ye, Zou and Drennan2006) suggest defect clustering up to a few nanometres. As a downside, the latter techniques only inspect a limited portion of the specimen, which may not be representative of the whole material.
$M_{{\rm Ce}}^{\prime} - V_0^{{\prime}{\prime}} - M_{{\rm Ce}}^{\prime}$
; however, such techniques only investigate the first few coordination shells of an atom. At odds with that, electron microscopy probes such as transmission electron microscopy (TEM), selected area electron diffraction (SAED), and electron energy loss spectroscopy (EELS) (Ou et al., Reference Ou, Mori, Ye, Zou and Drennan2006) suggest defect clustering up to a few nanometres. As a downside, the latter techniques only inspect a limited portion of the specimen, which may not be representative of the whole material.
To overcome some of those drawbacks, we recently exploited the pair distribution function (PDF) method to investigate doped ceria compounds upon doping with Gd (Scavini et al., Reference Scavini, Coduri, Allieta, Brunelli and Ferrero2012), Y (Coduri et al., Reference Coduri, Brunelli, Scavini, Allieta, Masala, Capogna, Fischer and Ferrero2012a, Reference Coduri, Scavini and Allieta2013a), and other rare earths (Coduri et al., Reference Coduri, Scavini, Allieta, Brunelli and Ferrero2012b, Reference Coduri, Scavini, Brunelli, Pedrazzin and Masala2014), also under fuel cell operating conditions (Coduri et al., Reference Coduri, Scavini, Brunelli and Masala2013b). The advantage of the PDF analysis over EXAFS lies in that the PDF function [i.e. G(r)] can yield information up to several hundreds of Ångstroms in real space, depending on the instrumental resolution. The PDF technique is also bulk-probing, as it relies on the scattered intensity from all the active volume of the sample. These capabilities are well suited to studying doped ceria, which brings in the so-called nanostructure problem (Billinge and Levin, Reference Billinge and Levin2007): the structure of concern is single-phase though perturbed on average by large, doping-induced static disorder, as inferred from the compositional evolution of atomic displacement parameters (Coduri et al., Reference Coduri, Brunelli, Scavini, Allieta, Masala, Capogna, Fischer and Ferrero2012a, Reference Coduri, Scavini and Allieta2012b, Reference Coduri, Scavini, Brunelli and Masala2013b; Scavini et al., Reference Scavini, Coduri, Allieta, Brunelli and Ferrero2012).
The key findings of the previous analyses cited above can be summarized as follows:
-
• The first coordination spheres in doped ceria samples can be pictured as the coexistence of dopant- and Ce-rich “droplets”, i.e. sub-nanometre regions with a distorted fluorite (CeO2) or C-type (dopant oxide) structure. The relative amount of the two kinds of droplets follows the compound stoichiometry (Coduri et al., Reference Coduri, Brunelli, Scavini, Allieta, Masala, Capogna, Fischer and Ferrero2012a, Reference Coduri, Scavini and Allieta2012b, Reference Coduri, Scavini, Brunelli and Masala2013b; Scavini et al., Reference Scavini, Coduri, Allieta, Brunelli and Ferrero2012).
-
• At intermediate dopant concentration (0.25 ≤ μ < 0.50), the dopant-rich droplets average out to C-type (dopant oxide-like) “domains”, the size of which spans some hundreds of Ångstroms (Coduri et al., Reference Coduri, Scavini and Allieta2013a).
-
• C-type domains grow in size with increasing dopant concentration, and connect through antiphase domain boundaries (APDB). APDBs, appearing in the Q-space patterns as the selective line broadening of superstructure reflections (Coduri et al., Reference Coduri, Scavini, Brunelli and Masala2013b), gradually fade with increasing dopant concentration.
In the frame of the investigation on doped ceria, this work specifically addresses the spatial correlation of defects by reviewing different structural models of the distribution of yttria-like defects in cerium oxide. The main structural features of Ce1−μ Y μ O2−μ/2 (CYO) are briefly recalled, pointing out the relationship between fluorite and C-type structures. In the latter, structural models are compared on the basis of their ability to fit to the experimental PDF curves for different compositions in ranges of interatomic distances spanning few hundreds of Ångstroms.
II. EXPERIMENTAL
A. Synthesis of CYO
Powdered samples of Y-doped ceria, CYO, were prepared according to the Pechini sol–gel method (Pechini, Reference Pechini1967), using Ce(NO3)3 and Y(NO3)3 (99 and 99.8%, Sigma-Aldrich) in due proportions. The precursor powders from the hydrothermal synthesis were uniaxially pressed with a 7 ton load to 2 mm-thick pellets, and sintered at 1173 K for 144 h with intermediate grindings. The obtained nominal compositions are μ = 0, 0.250, 0.313, 0.344, 0.438, 0.500, and 1.
B. X-ray diffraction data collection, processing and analysis
X-ray powder diffraction (XRPD) patterns were collected at the ID31 beamline (now ID22) of the ESRF, Grenoble. Isothermal measurements were carried out by setting T = 90 K. The X-ray wavelength was λ 1 = 0.35413(3) Å, and data were collected up to Q max = 29.4 Å−1, with Q = 4πsinθ/λ the wave-vector. The sample with μ = 0.500 was probed in a separate experimental run, using a wavelength λ 2 = 0.3099(2) Å, reaching Q max = 28.0 Å−1. The measured contribution of the empty capillary was subtracted from the PDF data.
The XRPD data were fitted using both the Rietveld method via the GSAS program (Larson and von Dreele, Reference Larson and Von Dreele2000) and its graphic interface EXPGUI (Toby, Reference Toby2001), and the Whole Powder Pattern Modelling (WPPM) approach as implemented by the PM2K code (Leoni et al., Reference Leoni, Confente and Scardi2006). G(r) curves were obtained through the PDFGetX2 program (Qiu et al., Reference Qiu, Thompson and Billinge2004), and modelled with the PDFgui program (Farrow et al., Reference Farrow, Juhás, Liu, Bryndin, Božin, Bloch, Proffen and Billinge2007).
As a formalism, we used the so called reduced PDF, G(r), obtained via the Fourier sine transform of the experimental total scattering function S(Q):
 $$\eqalign{& G\lpar r\rpar =4\pi r\lsqb {\rho \lpar r \rpar - {\rho_0}} \rsqb = \cr & \quad \displaystyle{2 \over \pi }\vint_0^\infty {Q\lsqb {S\lpar Q \rpar - 1} \rsqb \sin \lpar {Qr} \rpar dQ}}$$
$$\eqalign{& G\lpar r\rpar =4\pi r\lsqb {\rho \lpar r \rpar - {\rho_0}} \rsqb = \cr & \quad \displaystyle{2 \over \pi }\vint_0^\infty {Q\lsqb {S\lpar Q \rpar - 1} \rsqb \sin \lpar {Qr} \rpar dQ}}$$
where ρ(r) is the atomic number density function, which denotes the probability of finding an atom at a distance r from another atom, and ρ 0 is the average number density.
The r-dependent fits to the G(r) curves were obtained following to the so-called “box-car” refinement strategy: selected structural parameters were iteratively refined throughout 20 Å wide boxes covering a range up to 500 Å, depending on the model and the sample. Thus, the variable r in the plots stands for the centroid of the “box-car” refinement range.
The degree of accuracy of the analysis was evaluated by calculating the residual factor as follows:
 $${R_W}=\sqrt {\displaystyle{{\sum\nolimits_{i=1}^n {\omega \lpar {{r_i}} \rpar {{\lsqb {G_{{\rm obs}}^{} \lpar {{r_i}} \rpar - G_{{\rm calc}}^{} \lpar {{r_i}} \rpar } \rsqb }^2}} } \over {\sum\nolimits_{i=1}^n {\omega \lpar {{r_i}} \rpar G_{{\rm obs}}^2 \lpar {{r_i}} \rpar } }}}$$
$${R_W}=\sqrt {\displaystyle{{\sum\nolimits_{i=1}^n {\omega \lpar {{r_i}} \rpar {{\lsqb {G_{{\rm obs}}^{} \lpar {{r_i}} \rpar - G_{{\rm calc}}^{} \lpar {{r_i}} \rpar } \rsqb }^2}} } \over {\sum\nolimits_{i=1}^n {\omega \lpar {{r_i}} \rpar G_{{\rm obs}}^2 \lpar {{r_i}} \rpar } }}}$$
where
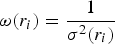 $\omega \lpar {r_i}\rpar =\displaystyle{1 \over {{\sigma ^2}\lpar {r_i}\rpar }}$
, σ(r
i
) being the standard deviation for a given r
i
distance value.
$\omega \lpar {r_i}\rpar =\displaystyle{1 \over {{\sigma ^2}\lpar {r_i}\rpar }}$
, σ(r
i
) being the standard deviation for a given r
i
distance value.
III. RESULTS AND DISCUSSION
The fluorite structure of CeO2 is described by the space group Fm-3m (225). In the CeO2 cell, Ce4+ occupies the (0,0,0) position and has eight coordinated O2− located in (1/4,1/4,1/4). The C-type structure of Y2O3, space group Ia-3 (206), can be seen as a 2 × 2 × 2 fluorite supercell whose origin is shifted by 1/4,1/4,1/4. In each octant, Y3+ occupies the sixfold coordinated 8b and 24d sites, O2− is in the 48e site, and two 16c sites are vacant. Table I reports atom coordinates and Wyckoff positions of the pertinent phases. Hereafter, the C-type setting is applied to the fluorite structure, so that all sites are unambiguously referred to for any composition. We take the fractional x-coordinate of the M2 site [hereafter x(M2)] as the reference parameter to discuss our data, as it is the only degree of freedom of the cationic substructure: when x(M2) = 0, the cation arrangement is the same as in fluorite, while it takes negative values in C-type.
Table I. Atom positions and corresponding Wyckoff symbols for the sites in fluorite, C-type, and fluorite with C-type setting. Notice that the 16c site is vacant in pure Y2O3.

A. Reciprocal space analysis
Table II outlines the results from Rietveld refinements of the XRPD patterns. All of the CYO samples were single-phase, and the average structure is Fm-3m fluorite for μ ≤ 0.250 and Ia-3 C-type for μ > 0.250. The cell parameter decreases with increasing yttrium concentration owing to the lower coordination and smaller ionic radius of Y3+ with respect to Ce4+ (Shannon and Prewitt, Reference Shannon and Prewitt1969; Coduri et al., Reference Coduri, Scavini, Allieta, Brunelli and Ferrero2012b).
Table II. Selected data from Rietveld refinements and WPPM best fits. For the μ = 0.250 sample the fluorite cell parameter a = 5.4032(5) Å has been doubled so as to report the value of the corresponding 2 × 2 × 2 supercell. U mean is the msd parameter averaged over all the sites in each structure.

As depicted in Figure 1, O2 oxygen vacancies (16c) and Ce/Y substitutional defects cause the x-coordinate of the C-type M2 cation to take intermediate values between those corresponding to the end members of the solid solution, that is x(M2) = 0 for CeO2, and x(M2)~(−0.032) for Y2O3. Such values are consistent with the distribution of M2 cations in two different chemical environments: (i) the centre of eight equidistant occupied oxygen sites [CeO2-like cuboids, Figure 1(a)], and (ii) an offset position because of two vacant O2 sites [Y2O3-like cuboids, Figure 1(c)].

Figure 1. (Colour online) (a) CeO2 cuboid, where the Ce4+ cation (M1 = M2) is centred in the eight-coordinated position; (b) Ce0.5Y0.5O1.75 cuboid, with 0.5 probability for a vacancy to be in the 16c site and a displaced M2 cation; (c) Y2O3 cuboid, in which every 16c site is empty. For each panel x(M2) yielded by the respective Rietveld fit is indicated.
The C-type structure of the samples with μ = 0.313, 0.344, and 0.438 is largely distorted with respect to reference Y2O3. The atomic mean square displacement (msd) peaks at μ = 0.438 (see Table II). Since measurements were performed at 90 K, the trend in the msd parameters is because of static disorder only.
Also recalled in Table II are the results of the WPPM best fits to XRPD patterns of the μ = 0.313, 0.344, and 0.438 samples, obtained using PM2K. Accurate fit of the superstructure peaks were attained only by accounting for antiphase domains with dopant-like structure. Ordering of defects starting at non-equivalent sites could have yielded unmerged domains splicing at APDBs. A similar mechanism was proposed for the Cu3Au alloy (Scardi and Leoni, Reference Scardi and Leoni2005).
A possible relationship between the APDB probability and the spatial extent of such domains prompted us to employ PDF analysis in order to measure the domain correlation distance.
B. Real-space analysis
The effect of composition on the cation environment is witnessed by the metal–metal (MM) nearest neighbour distances in Figure 2. At the lowest dopant concentration already, the single MM distance of CeO2 [Figure 2(a)] splits into a shorter (MMa~3.75 Å) and a longer distance (MMb~4.10 Å). The G(r) of all the samples was fitted in the range between 1.5 and 5 Å. With reference to μ = 0.250, 0.344, and 0.500 samples, Figures 2(b)–2(d) show the G(r) in the 3 < r < 5 Å range as fitted by fluorite (μ = 0.250) and C-type (μ = 0.344 and 0.500) long-range models. As to the μ = 0.250 sample, a fluorite phase could not reproduce the MMb peak. In fact, the MMb interatomic distance tallies with an atom pair involving the C-type M2 site [see Y2O3 in Figure 2(h)]. However, in the G(r) of the μ = 0.344 and 0.500 samples, the MMb peak is equally mismatched when a C-type phase is fitted into it.

Figure 2. (Colour online) Experimental (circles) PDF curves in the 3 < r < 5 Å range for samples with μ = 0.250, 0.344, and 0.500 composition fitted above (b–d) by the fluorite (green dashed line) and C-type (blue solid line) average models; below (e–g) by the two-phase model (red solid line), to which the partial contributions of the fluorite (green solid line) and C-type (blue dotted line) phases are also plotted. (a) Best fit based on the single-phase fluorite structure for CeO2 (solid line); (h) best fit based on the single-phase C-type structure for Y2O3 (blue solid line). Fit residuals are indicated in each frame (solid line below the fitted curves and right-hand side of the frame).
Either single phase fitted poorly the G(r) in this range, as neither of them could be adapted to fit G(r) peaks originating from the other phase. A reasonable solution was provided by a two-phase model, which considers the first atomic coordination shells as a combination of the fluorite and C-type chemical environments (Coduri et al., Reference Coduri, Scavini, Allieta, Brunelli and Ferrero2012b; Scavini et al., Reference Scavini, Coduri, Allieta, Brunelli and Ferrero2012). The fit by the two-phase model of the G(r) curves is depicted in Figures 2(e)–2(g) together with the partial contributions from the fluorite and C-type phases. An overview of the refined parameters is given in Table III (see “relaxed two-phase”). The change in the relative intensity and position of the MMa and MMb peaks upon increasing μ, i.e. the lesser occurrence of pairs involving Ce replaced by Y–M pairs is accounted for not only by the phase scale-factor parameters, but also by x(M2). At μ = 0.250 already, the value of x(M2) refined by the two-phase model is close to that in Y2O3 despite the fluorite average structure of this sample.
Table III. Overview of the structural assessed in this work with the respective box-car refinement parameters. Scale (f,c) stands for the relative phase fractions; a F and a C are the cell parameter of fluorite and C-type phases, respectively; U iso indicates the isotropic msd (when U iso is applied to two atoms, both share the same value); U ii is an anisotropic component of msd.

This limited spatial extent over which C-type and fluorite interatomic distances are described by the two-phase model was defined as “droplet” (Scavini et al., Reference Scavini, Coduri, Allieta, Brunelli and Ferrero2012; Coduri et al., Reference Coduri, Scavini and Allieta2013a). As a result of both the tiny coherence length and random orientation, droplets average out to zero in the reciprocal space and do not constitute a secondary phase in the XRPD patterns. Thus, using a two-phase model for describing the first few atomic distances is consistent with the observation in the diffraction patterns that every CYO sample is single-phase.
To find out how the dopant-like atomic environment relaxes out of the first coordination shell, C-type and fluorite contributions must be probed further in the r space for each sample. High-quality PDF data were needed to apply different models to the experimental G(r) on a wide r range. Figure 3(a) reports the G(r) up to 600 Å for the μ = 0.250, 0.313, 0.344, 0.438, and 0.500 samples. The slow falloff of the G(r) amplitude owes to the narrow instrumental resolution function of the ID31 diffractometer. Noise in the PDF data was minimized by increasing counting statistic thanks to: (i) long collection time, especially in the high Q range, (ii) low temperature of the experiment (90 K), which kept the Debye–Waller factor low.

Figure 3. (Colour online) Experimental G(r) curves up to r = 600 Å (left) for the CYO samples. Details of the curves in the range 395 < r < 400 Å are also shown (right). The dashed line is a guide to the eye tracing the peak position.
In the G(r) curves discussed, signal-to-noise ratio and peak resolution were satisfactory even at 400 Å. For instance, we could detect a shift in position, shape, and width of the two peaks of the doublet at ~397 Å depicted in Figure 3(b). The traces of this doublet upon increasing μ match the shortening of the interatomic distances and the increase in the DW factor at μ = 0.438 worked out in reciprocal space.
In the following paragraphs, structural parameters refined against G(r) in successive 20 Å ranges (box-car refinements) are discussed through their trends as a function of r. A list of the refined parameters for the models introduced is given in Table III.
As it yielded the best solution on the local scale, the two-phase model was tested on further interatomic distances in two different ways: (i) by keeping the positional degrees of freedom [cell parameters and x(M2)] fixed, as in the local scale (r < 5 Å) refinement, referred to as “unrelaxed two-phase”; (ii) by refining the positional degrees of freedom, referred to as “relaxed two-phase”. Hereafter we will call the refined scale-factor of the C-type phase as “C-fraction”.
Figure 4(a) shows the unrelaxed two-phase model fitted against the G(r) of the μ = 0.250 sample in the 5 < r < 15 Å range. It can be noted from the misshapen peaks and the high fit residual that such model becomes unreliable as soon as the fluorite and C-type distances are not resolved. Actually, for r > 10 Å, the G(r) vectors span over the length of a droplet, and thereby they cannot be decomposed into the distinct contributions from two different coordination environments. By letting also phase cell parameters and x(M2) be refined, the fit quality improved significantly [Figure 4(b)], hence, the r-dependent structure refinement was carried up to hundreds of Å out using the relaxed model.

Figure 4. (Colour online) Experimental (circles) and model fitted PDF curves in the 5 < r < 15 Å range for the sample with μ = 0.250 composition: (a) unrelaxed two-phase model; (b) relaxed two-phase model; (c) isotropic C-type model; (d) anisotropic C-type model; (e) fluorite model. Below each fitted curve the fit residual (green solid line) is shown along with its zero-point line (dotted line).
The trends of x(M2) and of C-fraction as a function of r obtained using the relaxed two-phase model are plotted in Figures 5(a) and 5(b). The cell parameters of the two phases correlate strongly when refined. Notably, C-fraction for the μ = 0.250 sample vanishes just over 100 Å, while the corresponding x(M2) value becomes unphysically negative. A similar observation can be made for the μ = 0.344 sample. In particular, in this case, the C-fraction cut-off is shifted to a higher r value (~210 Å), but it is poorly determined as its value declines to zero very slowly. Unlike this trend, in the sample μ = 0.313, x(M2) goes neatly to zero, while C-fraction is almost constant with r.

Figure 5. (Colour online) Plots of the refined parameters vs. r for the samples μ = 0.250, 0.313, 0.344, and 0.438 as from: (a–c) the relaxed two-phase model, (d) the isotropic C-type model. Horizontal solid lines in the plots indicate x(M2) ± 0.004 as a reference.
Despite the large fluctuation in the values because of high parameter correlation, such trend suggests a structural evolution from C-type to fluorite when increasing r in the samples μ = 0.250, 0.313, and 0.344. Indeed, the vanishing of either the C-fraction or x(M2) supports the only presence of the fluorite phase at high r. On the other hand, the μ = 0.438 sample shows no fluorite character even at 500 Å.
Bearing in mind that both the x(M2) and C-fraction parameters express the deviation from the fluorite structure, and that the two parameters exhibit a major negative correlation, we derived a C-type-to-fluorite order parameter by multiplying x(M2) by C-fraction through the whole refinement range. The x(M2) × C-fraction parameter, plotted in Figure 5(c), becomes zero at r~120, ~110, and ~200 Å, respectively, for μ = 0.250, 0.313, and 0.344. As expected, at μ = 0.438, the x(M2) × C-fraction parameter does not approach zero at any distance.
In the refinements against the relaxed two-phase model, the C-type cell parameter converged to a value twice the fluorite cell parameter. Therefore, a meaningful description of the structure could be achieved using a single C-type model [Figure 4(c)], thereby cutting the correlation between fluorite and C-type structural parameters. In the C-type model, site occupancy factors of Ce and Y atoms mirror the sample composition, and the 16c site is filled accordingly. As a result, the x(M2) parameter returned by the refinement was averaged over the two unalike positions of Ce and Y in their chemical environment, thus including the relative weights of the fluorite and C-type phases. While this was not satisfying on the local scale, it afforded a good fit at the nanometre scale.
Lower fit residual was achieved as compared with the relaxed two-phase model. The cell parameters evolve negligibly along r in all of the samples, and closely resemble the respective values from Rietveld refinements: a~10.807, ~10.787, ~10.780, and ~10.769 Å for μ = 0.250, 0.313, 0.344, and 0.438. No noteworthy correlations involving x(M2) were found and, therefore, the C-type domain size could be accurately estimated by only using the r-dependence of x(M2).
The trends of x(M2) vs. r yielded by the C-type model are plotted in Figure 5(d). For μ = 0.250, 0.313, and 0.344, x(M2) averages out to zero at r~120, ~140, and ~180 Å, respectively. On the contrary, μ = 0.438, x(M2) slopes in the range −0.014 < x(M2)< −0.010, but does not become zero within a range of 500 Å. This result points out that the C-type phase virtually achieves long-range coherence at μ = 0.438, whereas the same ordering is confined in 100–200 Å-sized domains at lower dopant concentrations. As can be noticed in Figure 5(d), the value of x(M2) within the domain gets more negative the higher the Y concentration in the sample. With increasing μ, fewer fluorite-like interatomic distances in the G(r) curve must be fitted by the C-type phase. Accordingly, the refined x(M2) parameter resembles more closely the value of x(M2) in Y2O3.
A possible improvement of the fitting model could be achieved by using a C-type phase along with anisotropic displacement parameters. Strong anisotropic static disorder is indeed typical of doped ceria compounds (Scavini et al., Reference Scavini, Coduri, Allieta, Brunelli and Ferrero2012; Coduri et al., Reference Coduri, Scavini, Brunelli and Masala2013b). Figure 4(d) shows the G(r) fit by this model in the 5 < r < 15 Å range for the μ = 0.250 composition.
Lower fit residual with respect to the previous C-type model (i.e., isotropic C-type) was attained using anisotropic thermal factors (i.e., anisotropic C-type). The refined cell parameter matched the value obtained by the isotropic C-type model for any composition and r range.
For the μ = 0.344 and 0.438 samples, the r-dependence of x(M2) parameter followed almost the same trend yielded by the isotropic model. Conversely, for the μ = 0.250 and 0.313 samples a lower x(M2) cut-off resulted (~40 Å) which totally disagrees with isotropic model results. This discrepancy is explained by the strong correlation between the x(M2) position and the U 11 parameter, which made this model inconsistent. Using the anisotropic C-type, the uncertainties on x(M2) were greater by an order of magnitude than those found using the isotropic C-type. For these reasons, we retained the isotropic C-type as the reference model for estimating domain size.
By inspecting the refined C-type phase of the samples μ = 0.250, 0.313, and 0.344, it must be pointed out that the uncertainty on x(M2) increased considerably as x(M2) approaches zero. Such an increase suggests that the G(r) might be fitted more accurately in the r range encompassing the domain boundary and further in r. Thus, a simple fluorite structural model was fitted against the G(r) up to 400 Å, so that the refined C-type and fluorite phases could be compared throughout r.
The difference, ∆R w [=R w (C-type) − R w (fluorite)], between the residual of fit by C-type and that by fluorite is plotted in Figure 6 for the four compositions μ = 0.250, 0.313, 0.344, and 0.438. The ∆R w difference is negative in the samples μ = 0.250, 0.313, and 0.344 within the respective domain size, indicating the better agreement of the C-type model with the observed G(r). The two fit residuals cancel each other out approximately at the domain boundary, then, ∆R w oscillates around zero in the further range of r, at which the refined phases virtually equate to each other. In fact, when a C-type phase was fitted against G(r) in an r range outside the C-type domain, x(M2) equalled 0 within the uncertainty. In the μ = 0.438 sample, the fit residuals never cross in the whole range of r, reflecting the trend of x(M2) vs. r.

Figure 6. (Colour online) Plot of the difference, ∆R w , between the fit residual as from the C-type isotropic model and from the fluorite model as a function of r for the samples μ = 0.250, 0.313, 0.344, and 0.438. On the left of the dashed vertical line the r-axis scale is expanded.
Finally, the respective best fits in the range of interatomic distances between 390 and 410 Å are displayed in Figure 7 for all the samples. Although the absolute G(r) intensity is decreased with respect to the local scale because of the instrumental contribution (see Figure 3), no noteworthy fit residual was observed. The randomly distributed difference curve accounts for some noise, at a minimum, though, thanks to the high counting statistics. The good quality of the fit further supports the goodness of our approach.

Figure 7. (Colour online) Experimental (circles) PDF curves in the 390 < r < 410 Å range for the samples (a) μ = 0.250, (b) μ = 0.313, (c) μ = 0.344, and (d) μ = 0.438 with the corresponding calculated profile (solid red line) based on the respective best fitting model. Below each fitted curve the fit residual (solid green line) is shown along with its zero-point line (dotted line).
IV. CONCLUSION
PDF analysis of CYO samples with μ = 0.250, 0.313, 0.344, and 0.438 composition was carried out. Several models conceiving the structural relationship between fluorite (CeO2-like) and C-type (Y2O3-like) phases were evaluated through box-car refinements: (i) a two-phase model (i.e., coexisting Y2O3-like and CeO2-like phases), in which cell parameters and the x(M2) cation coordinate were either fixed or free to refine; (ii) a single C-type phase, refining either isotropic or anisotropic displacement parameters; and (iii) a single fluorite phase.
The model best describing the G(r) curves up to high r, attaining both accurate fits and meaningful structural parameters, is a simple C-type phase with isotropic displacement parameters. Nanometre-sized dopant domains were determined as the range in real space in which the C-type phase best fitted the experimental G(r). Gradual transformation of C-type into fluorite was observed with increasing r in the μ = 0.250, 0.313, and 0.344 samples. Different models, however, generally agree in the estimate of the C-type domain sizes, that is, nearly 100–140 Å for μ = 0.250 and 0.313, nearly 180–200 Å for μ = 0.344, and in excess of 500 Å for μ = 0.438.
ACKNOWLEDGEMENTS
The authors acknowledge the European Synchrotron Radiation Facility for provision of beam time; they wish to thank Dr. A. N. Fitch (ESRF) for assistance in using the ID31 beamline and also Dr. P. Masala (Università degli Studi di Milano) for useful discussions.










