


Introduction
Analysis of impurities in ice cores can yield evidence of past climatic change. Deep ice cores collected from the interior of the Antarctic ice sheet have provided records of the global atmospheric aerosol over many millennia.
The Antarctic Peninsula offers an ideal area in which to investigate the relationship between climatic conditions and the aerosol preserved in ice. The peninsula occupies a unique geographical position as the only land mass cutting across the sub-Antarctic zone. Climatic records deduced from ice cores collected along the peninsula will correlate ice-core records from the interior of the continent with climatic data from South America. The high accumulation rate, together with the availability of long-term meteorological observations for the area, allows a comparison of climate and ice-core impurities which includes seasonal detail.
Aristarain and others (1982) have measured a range of major impurities in a core collected at James Ross Island, at the northern end of the peninsula (Fig.1). This paper describes preliminary results from two cores drilled farther south in the peninsula. The first, collected at Dolleman Island on the east coast, contains a record of the aerosol at a low-altitude site with a pseudo-continental climate which is dominated by the proximity of the Weddell Sea. In contrast, the second core was collected from the spine of the peninsula, where the fluctuations in the perennial sea ice should have a less significant effect on the balance of chemical species deposited in the snow.
Experimental Procedures
Figure 1 shows the location of the two drilling sites. In January 1981, a core was collected from a site near Gomez Nunatak on the Palmer Land plateau (74°01’S, 70°38’W; 1130 m a.s.l.). The 10 m temperature at this site was −17.32 ° C, and the average accumulation rate was 0.88 m water equivalent / a. A 75 mm diameter core was retrieved to a depth of 82.47 m, using an electro-mechanical drill loaned by NSF/PICO. Results for the upper 47.2 m are presented here. The second core (10.5 m) was collected on Dolleman Island (70°35.2’S, 60°55.5’W) in January 1986. The site was 1 km west of the summit of the ice dome, 398 m a.s.l. The 10 m temperature was −16.75°C and the average accumulation rate in the upper part of the core was 0.54 m water equivalent / a. This core had a diameter of 103 mm, and was retrieved using a Japanese 1LTS-130-GT electro-mechanical drill.

Fig. l. Map of the Antarctic Peninsula, showing core-drilling sites cited in this paper.
Great care was taken to avoid contamination during sampling and analysis of the core. Standard clean procedures were adopted in the field for the handling of both cores. Sections of core were wrapped in polyethylene sleeving, protected by cardboard tubes, and transported frozen to the UK for analysis.
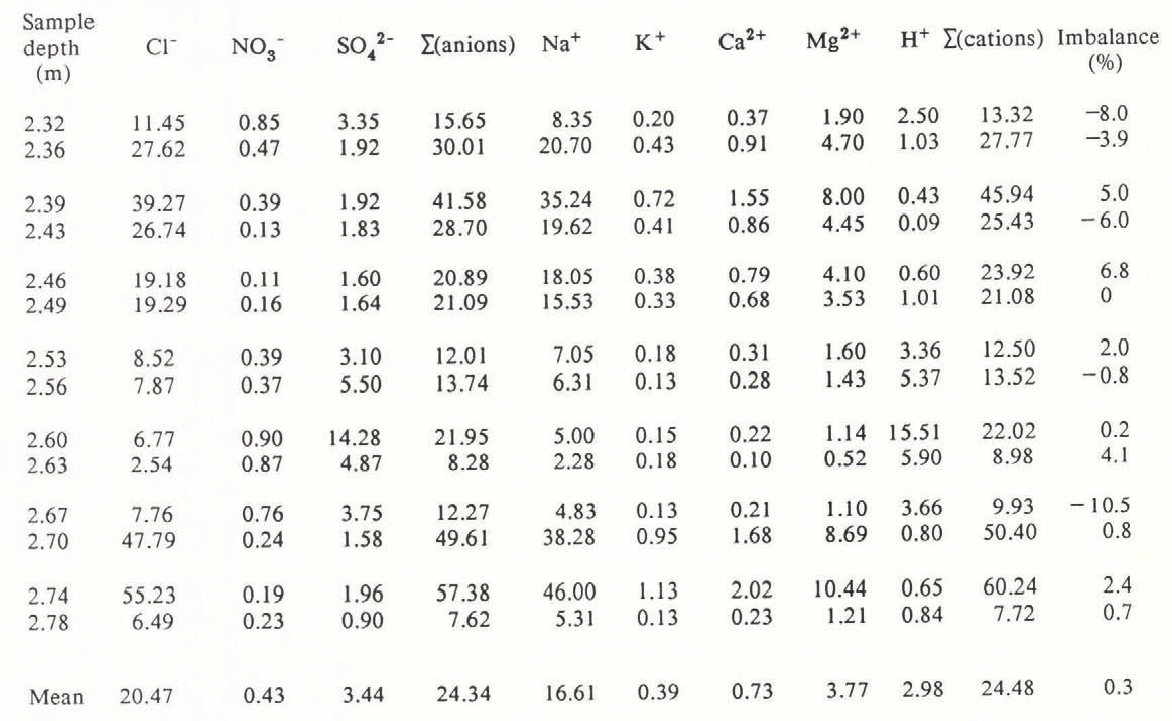
Table I. Ionic Concentrations (In µEq l−1) for the Major Anions and Cations from 1.5 Years’ Accumulation at Dolleman Island.
Mg2+ and Ca2+ values were calculated from the Na+ values, assuming sea-water ratios i.e. [Mg 2+]= 0.277 [Na+] and [Ca2+] = 0.044 [Na+]. The imbalance of cations to anions is expressed as a percentage of the sum of all ions; a negative value indicates a deficiency in the total cation value. NH4 + values are not included because of measurement difficulties (see text), but are <0.5µEq 1−1.
Sub-sampling of both cores was carried out in a cold-room at −20°C. Slices of the core were cut at a frequency of 7–30 samples per year’s accumulation, using a band saw whose blade was cleaned before each sampling session by repeatedly cutting a “core” of frozen ultra-pure water. Each slice was transferred immediately to a thermal sub-coring device on a laminar-flow clean-air bench within the cold-room. The device melts out an inner core of ice and the melt water is aspirated directly to a clean polyethylene bottle. This procedure prevents any contamination which may be present on the surface of the outer part from being incorporated into the sub-sample. (The diameter of the melting probe was 42 mm for the Gomez core and 66 mm for the Dolleman core.) The sub-sample was then taken directly to a laminar-flow work station, where it was analysed. All sample containers used in the analyses were cleaned by several 24 hour soaks in 18 Mohm ultra-pure water from a Millipore Milli-Q water system. Procedural blanks were included amongst the samples in order to assess the degree of contamination. The residual outer part of the core was melted in a sealed plastic bag and used for analysis of the stable oxygen-isotope ratio and the total beta decay. Dating of the core was by reference to the δ18O and total beta-decay profiles and is described by Peel and others (1988, this volume). The 47.2 m Gomez core spans 38 years of accumulation, whereas the 10.5 m Dolleman core covers 12 years.
Complete profiles of the anions Cl−, NO3 − and SO4 2− have been obtained for both cores. The cations Na+, K4−, NH.4+ and H+ were measured for one complete year of accumulation from the Dolleman core, to give an estimate of the ionic balance. All ions except H+ were measured by ion chromatography, using a Dionex model 2010i. An AS4A separator column, with standard carbonate/bicarbonate eluant, was used for Cl−, NO3 − and SO4 2−, whereas a CSI column with an HCl eluant was used for Na+, K+ and NH4 +. Analytical precision for these ions was better than 10% and the procedural blanks through all stages of the sampling and analytical procedure proved negligible at less than 0.05 µEq l−1 (micro-equivalents per litre) for all ions except NH4 +, which is discussed further below. Concentrator columns were used to pre-concentrate the snow samples before injection on to the separator columns: sample volumes were 2.5 ml for the anions and 8 ml for the cations. H+ was measured by acid titration (Legrand and others 1982) using a Radiometer Ion 85 system, a method which suppresses the weak acid contribution from the dissociation of carbonic acid. Precision by this method was ±0.2 µEq l−1. The calibration standards showed a blank of O.6 µEq l−1. The cause of this is unknown, but all samples have been corrected for it.
The ammonium ion proved very difficult to measure correctly, a point already made by Legrand and others (1984). The rate of uptake of NH3, from the laboratory air was so rapid as to swamp the NH4 + signal from the snow samples during the time taken to load the concentrator column. The source of NH3 was traced to a nearby reprography office. From the chromatograms it was possible to estimate the maximum value for NH4 + in the core as about 0,5 µEq l−1. This compares with the range of 0.07–0.19 µEq l−1 reported by Reference Legrand and DelmasLegrand and Delmas (1984) for a series of surface-snow samples. Thus the NH4 + results are not reported here, but they do not contribute significantly to the total ionic budget.
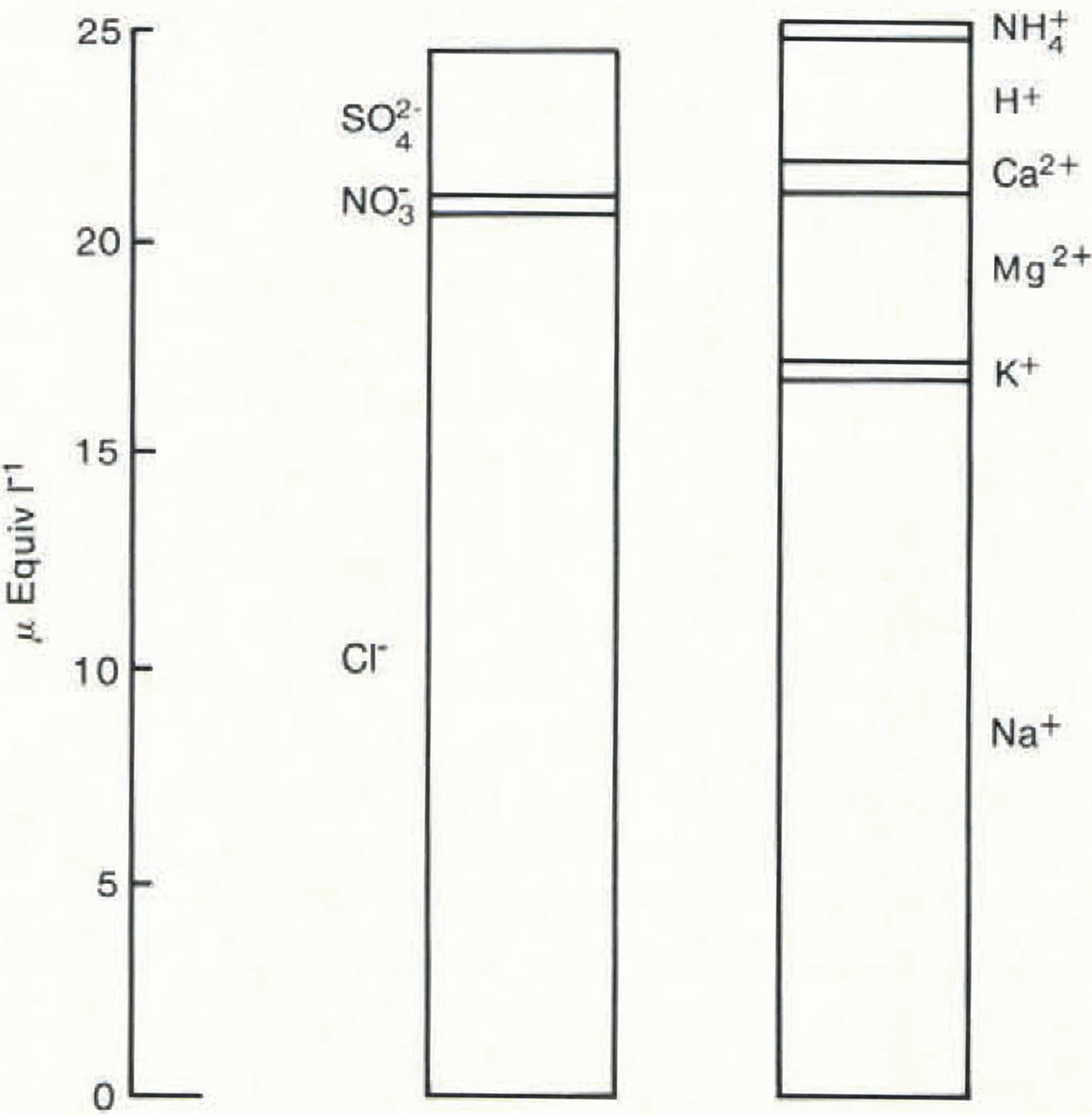
Fig. 2. The mean ionic balance for 1.5 years’ snowfall at Dolleman Island. The values for Ca2+ and Mg2+ were calculated from Na+; the NH4 + value is the estimated maximum.

Fig. 3. Comparison of the anion and oxygen-isotope data for the Gomez core, spanning the period 1942–80 (612 sample points). Winter horizons are shown at 5 year intervals. The 1964-65 total beta horizon is marked ** (from Peel and others 1988, this volume).
Results
Ionic budget
Table I and Figure 2 show the results of the ionic budget measured through an annual cycle of the Dolleman core. The 14 samples span the depths 2.32–2.78 m (water equivalent) and correspond approximately to snowfall for the period January 1979–July 1980. Mg2+ and Ca2+ have been calculated from the Na+ values, assuming that they are present in Antarctic snow in a sea-water ratio: i.e. Ca2+/Na+ = 0.044, Mg2+/Na+ = 0.227, expressed in equivalents per litre (from Walton Smith 1974). Reference BoutronBoutron (1982) showed that Mg2+ and Ca2+ derived from crustal sources are minor contributions to the ionic budget of Antarctic snowfall. The balance between cations and anions is very close (0.3% of the total ionic budget), although any contribution from NH4 + would add to the slight excess of cations. The individual imbalance for each sample is shown in Table I and each is within the analytical error inherent in the measurement of six species. The close balance of the mean ionic budget shows that our systematic errors are negligible. We therefore conclude that all significant ions, with the exception of NH4 +, have been accounted for.
The anion profiles
Figure 3 compares the anion and δ18O profiles for the Gomez core. Figure 4 shows similar profiles for the Dolleman core and the results are consistent with the coastal location of the site of this core. The high proportion of Na+ and Cl− in a near sea-water ratio indicates that the source of most of the aerosol precipitated at this site is sea salt. The profile for Cl− shows a strong seasonal signal, particularly prominent between 2 and 4 m water equivalent depth. We show below that the peak in Cl− corresponds to the late-summer/autumn period. During this time there is the maximum amount of open water in the Weddell Sea, and a south-easterly air stream provides a steady flux of sea-salt derived aerosol to the snowfall.

Fig. 4. Comparison of the anion and oxygen-isotope data for the Dolleman core, spanning the period 1973–85 (143 sample points). Winter horizons are shown at 5 year intervals.
In the Gomez core, the clear seasonal signal in Cl− is missing. The site lies at a higher altitude and at a greater distance from open sea water, hence a significant part of the sea-salt content of incoming air masses is deposited before they reach the site. This is reflected by the lower average Cl− value of 2.35 µEq l−1 for this core, compared to 11.76 µEq l−1 for the Dolleman core.
In contrast, the SO4 2− profile has a clear annual signal in both cores. The δ18O profile indicates that the maximum in SO4 2− occurs during the summer. The SO4 2− profile has proved sufficiently clear in its discrimination between summer and winter snowfall for us to recommend its use as a further aid to dating high-resolution ice-core records, in conjunction with the established techniques of stable-isotope and total beta-decay analysis.
Global pollution may contribute additional anions to the cores, in the form of the strong acids. During the period covered by the Gomez data, it has been shown (Neftel and others 1985, Mayewski and others 1986) that there has been a doubling of the SO4 2− and NO3 − concentrations in ice cores collected in Greenland, as a consequence of pollution in the Northern Hemisphere. We have calculated a simple linear regression for each anion over the 38 years of the Gomez data and found the slopes to be zero (<1% confidence level) in each case. We conclude therefore that the effect of anthropogenically derived SO4 2−, NO3 2− and Cl− on our data is negligible. (We have not yet tested our data for shorter-term climate-related trends.)
We have presented profiles here for the nitrate in the two cores, but our understanding of its source is still rudimentary and discussion of this data will be presented elsewhere. Reference Legrand and DelmasLegrand and Delmas (1986) recently concluded that Antarctic nitrate is contributed mainly by lightning at mid-latitudes, with possible modulation of concentrations by transport or deposition effects.
Annual cycles
To compare the seasonal signal in the anions we have calculated average annual-concentration profiles for each of the anions and for δ18O. The data for each year of accumulation, between depths corresponding to winter minima in the δ18O profile, were interpolated in order to give concentration values on a standard 24 sample points per annum. Each sampling point was then averaged over the sequence of complete years, to produce the average annual cycles shown in Figure 5. Calendar months have been assigned with reference to the isotopic minimum value which occurs, on average, in early August (±1 month) in this region (Peel and others 1988, this volume) and they assume a uniform snow-accumulation rate through the year.

Fig. 5. The mean annual cycle of the anions measured over 12 years for Dolleman Island and 38 years for Gomez Nunatak. Excess sulphate (excess SO4 2−) was calculated as [excess SO4 2−] = [SO4 2−] − 0.103 [Cl−]. Concentrations are in µEq l−1, except for δ18O, which is in ‰.
Two seasonal sources of sulphate are considered to be important (Reference Legrand, Aristarain and DelmasDelmas 1982): the primary source is derived from the SO4 2− found in sea salt, and a secondary source is the photochemically oxidized sulphurous gases derived from biogenic decay in the ocean, usually referred to as “excess sulphate”. To determine the contribution of the excess sulphate, we have calculated [excess SO4 2−] = [total SO4 2−] − 0.103 [Cl−] (in micro-equivalents). It is more usual to use Na+ as the sea-salt marker, since Cl− itself can occur as an excess over the sea-salt ratio (Reference Legrand and DelmasLegrand and Delmas 1984). We have not yet obtained a complete Na+ profile for either core. However, Table I shows that there is an excess of about 6% Cl− over the sea-water Cl−/Na+ ratio in this short section of the Dolleman Island core. This implies that the calculated excess SO4 2− is likely to be less than 1% lower than a value based on Na+ (rather than Cl−) at this site.
The annual cycle in excess SO4 2− shows a clear maximum in the late-February snowfall. If the mechanism for incorporation of non-sea-salt SO4 2− into the snowfall was dependent only upon the amount of sunlight penetrating to the troposphere, we would expect to see the maximum in the SO4 2− profile, to correspond to a date in late December. The observation that the maximum appears in early February may be explained in two ways: (i) the dating may be incorrect; (ii) the presence of non-sea-salt SO4 2− in the Antarctic aerosol is dependent both on sunlight to initiate the photochemical oxidation, and on the quantity of precursors available. We have rejected the first possibility because Peel and others (1988) have shown quite conclusively that the minima and maxima in the δ18O profile correspond to dates in early August and early February in cores from this part of the Antarctic. The second explanation requires the source of oxidation precursors to vary during the year. This could be related to the extent of open water around the peninsula, and perhaps to the degree of biological activity within the ocean. Both processes can be expected to lag the profile in air temperatures (and δ18O). We do not yet have sufficient evidence to show if either of these possibilities is likely.
In the Dolleman core, the chloride is at a maximum in iate-summer / autumn, and falls to a minimum in the late-winter / spring snowfall. This seems to be a clear indication that the incorporation of Cl− into the coastal aerosol is greatly dependent on the amount of open sea water available as a source.
The chloride annual cycle in the Gomez core is more complex. The 38 year profile shows little evidence of a seasonal periodicity in the influx of Cl− to the plateau site. The appearance in this profile of relatively high values of Cl− with a short duration of only one or two samples (representing 80–150 mm of snowfall) suggests that they are associated with discrete storm events. Averaged over 38 years, the annual cycle in Figure 5 implies that such events take place more frequently during the equinoxes. The autumnal equinox is associated also with minimum sea-ice coverage and this may account for the strong signal during March-May. On the average, the incidence of gales in this region is a factor of l½ greater during the period March-May than during December-February (personal communication from D.W.S. Limbert).
Conclusions
We have presented high-resolution profiles of Cl−, NO3 − and SO4 2− according to depth for two cores from the Antarctic Peninsula, from sites on the spine of the peninsula and the east coast, spanning 38 and 12 years respectively.
Our interpretation of the data is, at this stage, preliminary. We have shown that impurities in the east-coast core are dominated by the proximity of open water in the Weddell Sea, which shows increased accumulation of sea-salt derived aerosol in the late summer, whereas the site on the spine shows that the influx of sea salt is both lower in total and more erratic in timing.
Sulphate concentrations have been shown to be strongly seasonal in character, suggesting the influence of photo-chemical oxidation in the troposphere of sulphurous precursors derived from oceanic biogenic activity. The ease of discrimination between winter and summer values of the non-sea-salt derived sulphate makes it useful as a secondary method of dating high-resolution ice cores.
Acknowledgements
The authors thank the many members of the British Antarctic Survey who took part in the two drilling programmes. We are grateful to Dr H B Clausen and Dr N Gundestrup of the Geological Isotope Laboratory, University of Copenhagen, who carried out the oxygen-isotope analyses on both ice cores. The US Polar Ice Coring Office kindly loaned an electro-mechanical drill for the collection of the ice core from Palmer Land plateau. We thank in particular B Davison for his considerable efforts in assisting the cutting and sampling of the core in the UK, and A Reid, who carried out the acid-titration analyses.






