
Abstract
Ion migration in inorganic and hybrid organic-inorganic metal halide perovskites causes unusual phenomena in photovoltaic devices, such as current-voltage hysteresis and photoinduced phase transformations. It is now well established that metal halide perovskites are mixed ionic-electronic conductors and halide ions are mobile at room temperature. The effect of various physical stimuli like light, heat, pressure, and applied bias can affect the distribution and movement of ions. However, the impact of each stimulus is not understood as the measured response includes a combination of factors. We explore, using first-principles density functional theory (DFT), halide migration mediated by vacancies in model cubic phases of CsPbX3 (X = Cl, Br, I) and systematically control the electronic, chemical, and mechanical conditions. We assess the potential energy landscape for ion migration and how device-relevant conditions and compositional engineering can influence the physical behaviour of halide perovskites.
Similar content being viewed by others

1 Introduction
The crystallography of cesium lead halide perovskites dates back to the 1950s, when CsPbX3 (X = Cl, Br, I) crystals were determined to adopt the perovskites structure, and transitions to the ideal cubic phase at high temperature were reported [1]. In the 1970s, Hirotsu et al. found that the low-temperature phase transitions are associated with the condensation of zone boundary phonon modes related to the rotation of Pb\(\text {X}_6\) octahedra [2, 3]. Large thermal displacements are a feature of the cesium lead halide structures [4]. However, the atoms do not simply vibrate around their ideal crystallographic positions. Mizusaki et al. measured high ionic conductivity in CsPbX3 samples. They proposed the migration of X ion vacancies with an energy barrier of 0.25−0.39 eV [5]. The energy barrier is relatively small compared to the activation energy of vacancy migrations for other ionic compounds, e.g. 0.93 eV for oxygen migration is SrTi\(\text {O}_3\) [6]. Later studies confirmed these materials to be predominately halide ion conductors [7,8,9,10,11].
More recently, significant attention has been given to the study of organic-inorganic halide perovskites because of their high power conversion efficiencies in solar cells [12]. Ion migration is also an intrinsic property of these perovskites. There is growing experimental evidence that ion diffusion has critical impact on the long-term operation stability of metal halide materials and devices [13,14,15]. Several studies indicate the low formation energy of native defects in CsPbX3 due to the their weak chemical bond strength [16,17,18,19]. This corresponds to a high equilibrium defect density (\(10^{16}-10^{18} \mathrm {cm}^{-3}\)) [20,21,22] that supports ionic-electronic conduction [10, 22,23,24]. Haruyama et al. calculated iodine vacancy-assisted diffusion barriers of 0.32−0.45 eV [25]. Using both experimental data and theoretical calculations, Eames et al. proposed vacancy-assisted iodide transport with an energy barrier of 0.58−0.68 eV [23]. Yang et al. calculated an energy barrier for the vacancy-assisted diffusion of iodine ions of 0.26 eV [26]. Despite the range of values, which are sensitive to both the model setup and choice of Hamiltonian (e.g. exchange-correlation functional), there is qualitative agreement that the halide ions move through halide vacancy defect sites. However, since the diffusion profiles depend on direction and phase (perovskite tilt system), it is not easy to compare different studies directly.
Interestingly, the ion transport behavior also changes under device conditions. Illumination provokes different diffusion kinetics compared to dark conditions [27, 28]. Kim et al. reported evidence that illumination with energies above the bandgap increases the ion conductivity by orders of magnitude [29]. First-principles calculations agree that a large number of point defects favour ionized charge states (e.g. V\(_{\mathrm {I}}^+\)). [22] Light irradiation may promote the formation of metastable charge states due to the capture of photogenerated electrons and holes [30,31,32, or even the formation of metastable defect pairs [33].
There are additional contributions at play under operating conditions. The chemical bond between the lead and halogen (X = I, Br, Cl) largely determines the bandgap in these systems. The bandgap can be engineered to achieve high efficiency and stability by forming mixed-anion materials. Nevertheless, halide phase segregation happens under illumination for wider bandgap materials formed by mixing iodine and bromine on the X site. Halide ions redistribute to form a more stable phase to minimize the Gibbs free energy. A number of models have been reported to explain the thermodynamics and kinetics at this process [34, 35]. Changes in defect behaviour due to internal strains and external pressure have been reported [36, 37]. By applying external pressure to structure, the mobility of halide ions is altered, as tracked through the kinetics of Br/I phase separation. Internal strains, which is naturally produced as the crystal grows, has been reported to accelerate non-radiative losses [38, 39].
In this study, we focus on ion transport via charged halide vacancy defects in inorganic perovskites. A systemic investigation is performed to understand the factors impacting ion migration behaviour including mechanical, chemical, and electrical stimulii. We chose the cubic phase of lead halide perovskites as a model system to isolate and quantify competing effects and trends. We carried out first-principles density functional theory (DFT) calculations of the activation energies \(E_{a}\) of X ion migrations. We cover changes in the ion diffusion behavior as a function of: (a) charge state; (b) halide species; (c) hydrostatic strain. Finally, a set of guidelines are proposed for tuning ion transport in these materials.
2 Results and discussion
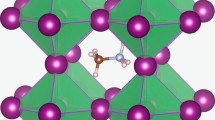
For each of the three CsPbX3 halides, we calculated the 2D potential energy surface for V\(_\mathrm{X}^{0}\) migration on a real-space grid. This approach provides access to both the energetic cost of ion migration and the corresponding pathway through the crystal. The diffusion pathway of an iodide vacancy, between equivalent halide sites, is shown in Fig. 1. The energy profiles are created by calculating the total energy of the supercell containing migrating ions along the diffusion path and identifying the appropriate saddle point. Vacancy migration reveals a curved path with the saddle point away from the neighboring B site. The curved path has been found from oxide-ion conduction in inorganic perovskites [40] and is also feature in halide perovskites crystal [23, 25].

Schematic of the cubic perovskite crystal structure (2 unit cells) is shown in the left panel. The vacancy-mediated diffusion pathway is indicated by the dashed arrow. Figure for Pb and I are colored black and purple. The right panel shows the calculated potential energy surface for iodine vacancy migration V\(_\mathrm{I}^{+}\) in CsPbI3 on a 41 \(\times\) 41 grid in a (100) plane using DFT (PBEsol)
2.1 Charge state
Vacancy mediated defect diffusion will be influenced by the charge state of the species, which depends on their formation energy. Under equilibrium conditions where the Fermi level is close to the centre of the band gap, the +1 charge state is favoured for halide vacancies in CsPbX3 [16,17,18,19]. When formed in charge-neutral combinations through Schottky or Frenkel ionic disorder, these charged defects have low energy to form, thus enabling high concentrations [22, 41].
Here we considered other possible charge states of V\(_\mathrm{X}\) which could be formed by illumination (carrier capture) or device conditions (Fermi level changes). To study ion migration behavior under light illumination, the calculations are repeated for each defect in different charge states. We note that while the negative and positive charges have an even number of electrons (closed shell), the neutral charge state has an odd number that requires spin-polarised calculations.
The activation energies for the transport of three X vacancies in different charge states (−, 0, +) are listed in Table 1. The corresponding diffusion path considered is illustrated in Fig. 1. The barrier height of the V\(_\mathrm{X}^{+}\) migration corresponds to the lowest saddle point between the minimum energy configurations. The activation energy increased as the charge state of the defect was reduced, and the same trend was observed across all halide species. e.g. from 0.34 eV (V\(_\mathrm{I}^{+}\)) to 0.50 eV (V\(_\mathrm{I}^{-}\)). This trend, under electron rich conditions, is related to the change in Coulomb repulsion due to the partial occupancy of the vacancy site by one (charge state 0) or two (charge state −) electrons. These findings can be linked to a reduction in mobility when defects trap charge, which can facilitate electron-hole recombination [42]. However, we note that other factors that could effect ion conductivity under illumination, such as the change in defect concentrations through photolysis [43].
2.2 Halide species
We next consider the relative stability and diffusion behavior of different halide species. The activation energy for iodide, bromide and chloride migration is 0.36 eV, 0.35 eV and 0.35 eV, respectively (Fig. 2). These values are very similar and must contain a cancellation of energy terms. While iodide has the largest ionic radius, it is also the most polarizable species. The longest interatomic separation for the lead iodide bond (\(\text {r}_{\text {Pb}-\text {I}}\) = 3.14 Å), compared to bromide (2.94 Å) and chloride (2.81Å), is associated with the weakest bond strength [44]. There is a further difference in the dielectric screening: the high-frequency dielectric constant increases as a function of the halide mass due to the decrease in bandgap [45, 46]. This effect alone should reduce the activation energy for the motion of charged defects down the series.

Calculated potential energy surface for vacancy-assisted V\(_\mathrm{X}^{+}\) halide migration in CsPbX3. This is the lowest energy path extracted from the 2D potential energy surface. The corresponding barriers are given in Table 1
High device performance with thermal and compositional stability has been observed for certain mixed halide perovskites [47, 48]. Here we consider the dilute incorporation of Br in CsPbI3 (i.e. a Cs8Pb8I22Br1 supercell) to compare iodide and bromide migration in comparison to CsPbI3 (Fig. 3). The energy barrier for Br to overcome in the vicinity of the vacancy is reduced by 0.2 eV compared to the pure iodide structure. Bromide has a smaller size, which provides more space for migration in an iodide structure, consistent with other reports [49]. The barrier undergoes a mild increase by 0.03 eV for I migration close to the Br substitution. The local strain caused by the shorter Pb−Br−Pb bonds is expected to be the dominant factor here.

Diffusion models considered in mixed halide systems. Pathway for iodine vacancy migration (purple circle) with: a a bromine ion in Cs8Pb8I22Br1 (reduced by 0.20 eV); b iodine ion migration in Cs8Pb8I22Br1 (increased by 0.03 eV); c reference iodine ion migration in pure CsPbI3
2.3 Hydrostatic strain
Strain can be caused in halide perovskite films by substrate effects, compositional inhomogeneity, or from thermal processing [50]. For instance, it can arise from the mismatched thermal expansion of the film and substrates. Large thermal expansion coefficients have been reported for these soft crystals [51]. Moreover, rather than being fixed, strain has been found to evolve under visible light illumination [52, 53]. While the understanding of strain distributions for crystals and films continues to evolve in the community, we note that the effect of pure hydrostatic pressure has been experimentally probed for a range of metal halide perovskites [37, 54].
Here, we simulated the role of mechanical perturbations using a model of CsPbX3 that is subjected to uniform expansion and compression. Calculations were performed at effective hydrostatic pressures ranging from − 2.0 (dilation) to 2.0 GPa (compression). All types of charge states and halide species were considered. The mapping between cell volume (V) and pressure (P) was performed using calculated bulk moduli of 17.34 GPa (CsPbI3), 21.70 GPa (CsPbBr3) and 24.76 GPa (CsPbCl3) using the standard relationship \(B_{0}=-V_{0} \frac{d P}{d V}\). The bulk moduli were obtained from fitting the energy-volume data to the third-order Birch-Murnaghan equation-of-state [55].

Change in activation energy for halide diffusion in CsPbX3 as a function of defect charge state and hydrostatic pressure. Negative pressure corresponding to a unit cell expansion. Points for Cl, Br and I are colored blue, green, coral
The energy barrier of halide ion migration increases linearly across the pressure range considered (Fig. 4). When the interatomic separations are decreased, there is less available space for the diffusing species, and the activation energy is increased. Similar slopes are found in each case, but some non-linearities appear owing to the compounding of electrostatic and steric factors. We note that similar barrier heights have been reported by by Smolders et al. [54]. for CsPbBr3, but a distinct change in anistropic diffusion behaviour was predicted above 1 GPa due to static octahedral tilting patterns that emerge.
3 Conclusions
Ion migration is a fundamental process that contributes to the unusual behaviour of metal halide perovskite materials and devices. Solid-state ion transport is facilitated by charged defects and thus intimately connected with charge-carrier recombination, transport rates, electrical instability, and thereby both device efficiency and lifetime [56, 57]. In general, ion migration should be suppressed in high-performance photovoltaic devices.
To understand the role of various stimuli, we separately simulated the effect of electrical, chemical, and mechanical changes on vacancy-mediated halide diffusion in CsPbX3. We found that the activation energies for vacancy-mediated diffusion in the stable charge states (V\(_\mathrm {I}^+\), V\(_{\mathrm {Br}}^+\), V\(_{\mathrm {Cl}}^+\)) are similar in the three halides. Our analysis showed that excess electrons increase the barrier for ion diffusion in each case. The same change is observed for materials placed under hydrostatic pressure (reduction in the unit cell volume).
A degree of freedom beyond the choice of halide, is the A-site cation. By changing the ratio of caesium, methylammonium, and formamidinium, the unit cell volume can be altered changing the diffusion kinetics [36]. The introduction of larger species such as dimethylammonium can further perturb the order of the crystal [58]. The leading effect of altering the A-site composition will be the unit cell volume, while the change in local structure and strains will also be important, especially for multi-cation mixtures. Indeed, the A-site cation can influence the kinetics and thermodynamics [59]. Recent research has shown that cation mixing can control the defect density to achieve stable and efficient photovoltaic devices [60].
While here we considered diffusion in the model cubic crystals, in reality, other effects can contribute to the physical response of these materials. The perovskite polymorphs accessible at room temperature vary between halides, resulting in distinct octahedral tilting patterns that can exhibit dynamic spatial fluctuations that will alter the diffusion behaviour. Beyond bulk factors, surfaces and extended defects such as grain boundaries will also influence the behaviour in polycrystalline films and devices, which should be considered in future studies. A deeper understanding of defect transport, trapping and accumulation is critical to improving the resistance of halide perovskites to environmental conditions.
4 Computational methods
4.1 Structure model
The cubic crystal structure of CsPbX3 (X = I, Br, Cl) is used, which corresponds to space group Pm3m. We considered ion transport in a 2 \(\times\)2 \(\times\)2 supercell which contains 40 atoms. The optimized unit cell lattice constants are 6.25 Å, 5.87 Å, and 5.62 Å for CsPbI3, CsPbBr3, and CsPbCl3, respectively. Based on the supercell expansion, one X ion is removed to obtain the vacancy defects, which corresponds to a defect fraction of 1/24. No internal degrees of freedom are considered. The individual jump distance of the halide ion vacancy is a/\({\sqrt{2}}\) and it has eight equivalent neighbors in three dimensions.
4.2 Density functional theory
The underlying total energies were calculated from DFT using VASP [61, 62], with the projector augmented-wave (PAW) [63] method. The Perdew-Burke-Ernzerhof exchange-correlation functional revised for solids (PBEsol) [62] was used to optimize structure. For all PBEsol calculations, the plane-wave kinetic energy cutoff was set to 700 eV, while convergence criteria of \(10^{-5}\) eV and \(10^{-2}\) eVÅ\(^{-1}\) for total energy and forces on each atoms, respectively, were employed.
The halide vacancies were modelled within periodic boundary conditions using the supercell approach [64]. For the positively and negatively charged defects, a homogeneous background charge was included to ensure charge neutrality and convergent electrostatic summations. The neutral halide vacancies result in an odd-electron count. Spin-polarised calculations were therefore performed, and this was found to be essential to describe the ground-state behaviour of V\(_\mathrm{Br}\) and V\(_\mathrm{Cl}\). These display localised F-centre like states, while the wavefunction associated with V\(_\mathrm{I}\) is more delocalised with no spin polarisation. As this study concerns relative energies for different defect configurations, no charge state correction schemes were applied. The low defect charge states considered (+/0/-) and supercell dimensions of \(> 10\) Å in each direction ensure that finite-size corrections are small in magnitude [65].
Data availability
The crystal structure files for the defective materials, and associated processing scripts, have been made available in an on-line repository at https://doi.org/10.5281/zenodo.6912860.
References
Møller CK. Crystal structure and photoconductivity of caesium plumbohalides. Nature. 1958;182:1436–1436.
Hirotsu S. Experimental studies of structural phase transitions in CsPbCl3. J Phys Soc Japan. 1971;31:552–60.
Hirotsu S, Harada J, Iizumi M, Gesi K. Structural phase transitions in CsPbBr3. J Phys Soc Japan. 1974;37:1393–8.
Sakata M, Nishiwaki T, Harada J. Neutron diffraction study of the structure of cubic CsPbBr3. J Phys Soc Japan. 1979;47:232–3.
Mizusaki J, Arai K, Fueki K. Ionic conduction of the perovskite-type halides. Solid State Ionics. 1983;11:203–11.
Schie M, Marchewka A, Müller T, De Souza RA, Waser R. Molecular dynamics simulations of oxygen vacancy diffusion in SrTi\(\text{O}_3\). J Phys Condens Matter. 2012;24:485002.
Narayan RL, Suryanarayana S. Transport properties of the perovskite-type halides. Mater Lett. 1991;11:305–8.
Hayashi H, Inaba H, Matsuyama M, Lan N, Dokiya M, Tagawa H. Structural consideration on the ionic conductivity of perovskite-type oxides. Solid State Ionics. 1999;122:1–15.
Yamada K, Isobe K, Tsuyama E, Okuda T, Furukawa Y. Chloride ion conductor \(\text{CH}_3\text{NH}_3\text{GeCl}_3\) studied by Rietveld analysis of X-ray diffraction and 35Cl NMR. Solid State Ionics. 1995;79:152–7.
Yang T-Y, Gregori G, Pellet N, Grätzel M, Maier J. The significance of ion conduction in a hybrid organic-inorganic lead-iodide-based perovskite photosensitizer. Angew Chem Int Ed. 2015;54:7905–10.
Yuan Y, Huang J. Ion migration in organometal trihalide perovskite and its impact on photovoltaic efficiency and stability. Acc Chem Res. 2016;49:286–93.
NREL, Best research-cell efficiency chart @ONLINE. 2021; https://www.nrel.gov/pv/cell-efficiency.html
Evarestov RA, Senocrate A, Kotomin EA, Maier J. First-principles calculations of iodine-related point defects in CsPb\(\text{I}_3\). Phys Chem Chem Phys. 2019;21:7841–6.
Evarestov R, Kotomin E, Senocrate A, Kremer R, Maier J. First-principles comparative study of perfect and defective CsPbX3 (X= Br, I) crystals. Phys Chem Chem Phys. 2020;22:3914–20.
Kim GY, Senocrate A, Wang Y-R, Moia D, Maier J. Photo-Effect on Ion Transport in Mixed Cation and Halide Perovskites and Implications for Photo-Demixing. Angew Chem Int Ed. 2021;60:820–6.
Li Y, Zhang C, Zhang X, Huang D, Shen Q, Cheng Y, Huang W. Intrinsic point defects in inorganic perovskite CsPb\(\text{I}_3\) from first-principles prediction. Appl Phys Lett. 2017;111:162106.
Kang J, Wang L-W. High defect tolerance in lead halide perovskite CsPb\(\text{Br}_3\). J Phys Chem Lett. 2017;8:489–93.
Sebastian M, Peters J, Stoumpos C, Im J, Kostina S, Liu Z, Kanatzidis MG, Freeman AJ, Wessels B. Excitonic emissions and above-band-gap luminescence in the single-crystal perovskite semiconductors CsPb\(\text{Cl}_3\) and CsPb\(\text{ Cl}_3\). Phys Rev B. 2015;92:235210.
Kye Y-H, Yu C-J, Jong U-G, Ri K-C, Kim J-S, Choe S-H, Hong S-N, Li S, Wilson JN, Walsh A. Vacancy-driven stabilization of the cubic perovskite polymorph of CsPb\(\text{I}_3\). J Phys Chem C. 2019;123:9735–44.
Stranks SD, Burlakov VM, Leijtens T, Ball JM, Goriely A, Snaith HJ. Recombination kinetics in organic-inorganic perovskites: excitons, free charge, and subgap states. Phys Rev Appl. 2014;2:034007.
Draguta S, Thakur S, Morozov YV, Wang Y, Manser JS, Kamat PV, Kuno M. Spatially non-uniform trap state densities in solution-processed hybrid perovskite thin films. J Phys Chem Lett. 2016;7:715–21.
Walsh A, Scanlon DO, Chen S, Gong X, Wei S-H. Self-regulation mechanism for charged point defects in hybrid halide perovskites. Ange Chem. 2015;127:1811–4.
Eames C, Frost JM, Barnes PR, O’regan BC, Walsh A, Islam MS. Ionic transport in hybrid lead iodide perovskite solar cells. Nat Commun. 2015;6:1–8.
Senocrate A, Moudrakovski I, Kim GY, Yang T-Y, Gregori G, Grätzel M, Maier J. The nature of ion conduction in methylammonium lead iodide: a multimethod approach. Angew Chem Int Ed. 2017;56:7755–9.
Haruyama J, Sodeyama K, Han L, Tateyama Y. First-principles study of ion diffusion in perovskite solar cell sensitizers. J Am Chem Soc. 2015;137:10048–51.
Yang J-H, Yin W-J, Park J-S, Wei S-H. Fast self-diffusion of ions in \(\text{CH}_3\text{NH}_3\text{PbI}_3\): the interstiticaly mechanism versus vacancy-assisted mechanism. J Mater Chem A. 2016;4:13105–12.
Xing J, Wang Q, Dong Q, Yuan Y, Fang Y, Huang J. Ultrafast ion migration in hybrid perovskite polycrystalline thin films under light and suppression in single crystals. Phys Chem Chem Phys. 2016;18:30484–90.
Zhao Y-C, Zhou W-K, Zhou X, Liu K-H, Yu D-P, Zhao Q. Quantification of light-enhanced ionic transport in lead iodide perovskite thin films and its solar cell applications. Light. 2017;6:e16243–e16243.
Kim GY, Senocrate A, Yang T-Y, Gregori G, Grätzel M, Maier J. Large tunable photoeffect on ion conduction in halide perovskites and implications for photodecomposition. Nat Mater. 2018;17:445–9.
Zhang X, Shen J-X, Van de Walle CG. First-principles simulation of carrier recombination mechanisms in Halide Perovskites. Adv Energy Mater. 2020;10:1902830.
Whalley LD, van Gerwen P, Frost JM, Kim S, Hood SN, Walsh A. Giant huang-rhys factor for electron capture by the iodine intersitial in perovskite solar cells. Soc J Am Chem. 2021. https://doi.org/10.1021/jacs.1c03064.
Keeble DJ, Wiktor J, Pathak SK, Phillips LJ, Dickmann M, Durose K, Snaith HJ, Egger W. Identification of lead vacancy defects in lead halide perovskites. Nat Commun. 2021;12:1–7.
Mosconi E, Meggiolaro D, Snaith HJ, Stranks SD, De Angelis F. Light-induced annihilation of Frenkel defects in organo-lead halide perovskites. Energy Environ Sci. 2016;9:3180–7.
Hoffman JB, Schleper AL, Kamat PV. Transformation of sintered cspbbr3 nanocrystals to cubic CsPbI3 and gradient CsPbBrxI3–x through halide exchange. J Am Chem Soc. 2016;138:8603–11.
Yoon SJ, Kuno M, Kamat PV. Shift happens. How halide ion defects influence photoinduced segregation in mixed halide perovskites. ACS Energy Lett. 2017;2:1507–14.
Muscarella LA, Hutter EM, Wittmann F, Woo YW, Jung Y-K, McGovern L, Versluis J, Walsh A, Bakker HJ, Ehrler B. Lattice compression increases the activation barrier for phase segregation in mixed-halide perovskites. ACS Energy Lett. 2020;5:3152–8.
Hutter EM, Muscarella LA, Wittmann F, Versluis J, McGovern L, Bakker HJ, Woo Y-W, Jung Y-K, Walsh A, Ehrler B. Thermodynamic stabilization of mixed-halide perovskites against phase segregation. Cell Rep Phys Sci. 2020;1:100120.
Jones TW, Osherov A, Alsari M, Sponseller M, Duck BC, Jung YK, Settens C, Niroui F, Brenes R, Stan CV, Li Y. Lattice strain causes non-radiative losses in halide perovskites. Energy Environ Sci. 2019;12:596–606.
Doherty TA, Nagane S, Kubicki DJ, Jung YK, Johnstone DN, Iqbal AN, Guo D, Frohna K, Danaie M, Tennyson EM, Macpherson S. Stabilized tilted-octahedra halide perovskites inhibit local formation of performance-limiting phases. Science. 2021;374:1598–605.
Cherry M, Islam MS, Catlow C. Oxygen ion migration in perovskite-type oxides. J Solid State Chem. 1995;118:125–32.
Meggiolaro D, Mosconi E, De Angelis F. Formation of surface defects dominates ion migration in lead-halide perovskites. ACS Energy Lett. 2019;4:779–85.
deQuilettes DW, Frohna K, Emin D, Kirchartz T, Bulovic V, Ginger DS, Stranks SD. Charge-carrier recombination in halide perovskites: focus review. Chem Rev. 2019;119:11007–19.
Walsh A, Stranks SD. Taking control of ion transport in halide perovskite solar cells. ACS Energy Lett. 2018;3:1983–90.
Pauling L. The nature of the chemical bond. Ithaca: Cornell University Press; 1960. p. 260.
Poglitsch A, Weber D. Dynamic disorder in methylammoniumtrihalogenoplumbates (II) observed by millimeter-wave spectroscopy. J Chem Phys. 1987;87:6373–8.
Sendner M, Nayak PK, Egger DA, Beck S, et al. Optical phonons in methylammonium lead halide perovskites and implications for charge transport. Mater Horiz. 2016;3:613–20.
Sutton RJ, Eperon GE, Miranda L, Parrott ES, Kamino BA, Patel JB, et al. Bandgap-tunable cesium lead halide perovskites with high thermal stability for efficient solar cells. Adv Energy Mater. 2016;6:1502458.
Wang H, Bian H, Jin Z, Zhang H, Liang L, Wen J, Wang Q, Ding L, Liu SF. Cesium lead mixed-halide perovskites for low-energy loss solar cells with efficiency beyond 17%. Chem Mater. 2019;31:6231–8.
McGovern L, Grimaldi G, Futscher MH, Hutter EM, Muscarella LA, Schmidt MC, Ehrler B. Reduced barrier for ion migration in mixed-halide perovskites. ACS Appl Energy Mater. 2021. https://doi.org/10.1021/acsaem.1c03095.
Liu D, Luo D, Iqbal AN, Orr KW, Doherty TA, Lu Z-H, Stranks SD, Zhang W. Strain analysis and engineering in halide perovskite photovoltaics. Nat Mater. 2021;20:1337–46.
Jacobsson TJ, Schwan LJ, Ottosson M, Hagfeldt A, Edvinsson T. Determination of thermal expansion coefficients and locating the temperature-induced phase transition in methylammonium lead perovskites using x-ray diffraction. Inorg Chem. 2015;54:10678–85.
Zhao J, Deng Y, Wei H, Zheng X, Yu Z, Shao Y, Shield JE, Huang J. Strained hybrid perovskite thin films and their impact on the intrinsic stability of perovskite solar cells. Sci Adv. 2017;3:eaao5616.
Zhu C, Niu X, Fu Y, Li N, Hu C, Chen Y, He X, Na G, Liu P, Zai H, Ge Y. Strain engineering in perovskite solar cells and its impacts on carrier dynamics. Nat Commun. 2019;10:1–11.
Smolders TJ, Walker AB, Wolf MJ. 3D-to-2D transition of anion vacancy mobility in CsPb\(\text{ Br}_3\) under hydrostatic pressure. J Phys Chem Lett. 2021;12:5169–77.
Birch F. Finite elastic strain of cubic crystals. Phys Rev. 1947;71:809–24.
Ball JM, Petrozza A. Defects in perovskite-halides and their effects in solar cells. Nat Energy. 2016;1:1–13.
Park N-G. Research direction toward scalable, stable, and high efficiency perovskite solar cells. Adv Energy Mater. 2020;10:1903106.
Simenas M, Balciunas S, Wilson JN, Svirskas S, Kinka M, Garbaras A, Kalendra V, Gagor A, Szewczyk D, Sieradzki A, Maczka M. Suppression of phase transitions and glass phase signatures in mixed cation halide perovskites. Nat Commun. 2020;11:1–9.
Saliba M, Matsui T, Seo JY, Domanski K, Correa-Baena JP, Nazeeruddin MK, Zakeeruddin SM, Tress W, Abate A, Hagfeldt A, et al. Cesium-containing triple cation perovskite solar cells: improved stability, reproducibility and high efficiency. Energy Environ Sci. 2016;9:1989–97.
Ceratti DR, Cohen AV, Tenne R, Rakita Y, Snarski L, Jasti NP, Cremonesi L, Cohen R, Weitman M, Rosenhek-Goldian I, Kaplan-Ashiri I. The pursuit of stability in halide perovskites: the monovalent cation and the key for surface and bulk self-healing. Mater Horiz. 2021;8:1570–86.
Kresse G, Furthmüller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys Rev B. 1996;54:11169.
Perdew JP, Ruzsinszky A, Csonka GI, Vydrov OA, Scuseria GE, Constantin LA, Zhou X, Burke K. Restoring the density-gradient expansion for exchange in solids and surfaces. Phys Rev Lett. 2008;100:136406.
Kresse G, Joubert D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys Rev B. 1999;59:1758.
Payne MC, Teter MP, Allan DC, Arias T, Joannopoulos AJ. Iterative minimization techniques for ab initio total-energy calculations: molecular dynamics and conjugate gradients. Rev Modern Phys. 1992;64:1045.
Freysoldt C, Neugebauer J, Van de Walle CG. Electrostatic interactions between charged defects in supercells. Phys Status Solidi B. 2011;248:1067–76.
Acknowledgements
We thank Ju-Lee Kim for coding assistance and Zhenzhu Li for useful suggestions. Computational resources have been provided by the KISTI Supercomputing Center (KSC-2020-CRE-0063). Y.W. was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (No. 2018R1C1B6008728). We are grateful to the UK Materials and Molecular Modelling Hub for computational resources, which is partially funded by EPSRC (EP/P020194/1 and EP/T022213/1).
Author information
Authors and Affiliations
Contributions
The following author contributions were assigned following the CRediT system. YWW: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Visualization, Writing—original draft, Writing—review & editing. YKJ: Investigation, Methodology, Writing—review & editing. GYK: Formal Analysis, Writing—review & editing. SK: Methodology, Formal Analysis, Writing—review & editing. AW: Conceptualization, Methodology, Supervision, Formal Analysis, Writing—review & editing. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Woo, Y.W., Jung, YK., Kim, G.Y. et al. Factors influencing halide vacancy transport in perovskite solar cells. Discov Mater 2, 8 (2022). https://doi.org/10.1007/s43939-022-00029-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s43939-022-00029-z