
Abstract
SARS-CoV-2 pandemic is the big issue of the whole world right now. The health community is struggling to rescue the public and countries from this spread, which revives time to time with different waves. Even the vaccination seems to be not prevents this spread. Accurate identification of infected people on time is essential these days to control the spread. So far, Polymerase chain reaction (PCR) and rapid antigen tests are widely used in this identification, accepting their own drawbacks. False negative cases are the menaces in this scenario. To avoid these problems, this study uses machine learning techniques to build a classification model with higher accuracy to filter the COVID-19 cases from the non-COVID individuals. Transcriptome data of the SARS-CoV-2 patients along with the control are used in this stratification using three different feature selection algorithms and seven classification models. Differently expressed genes also studied between these two groups of people and used in this classification. Results shows that mutual information (or DEGs) along with naïve Bayes (or SVM) gives the best accuracy (0.98 ± 0.04) among these methods.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
SARS-CoV-2 is the current health issue of the globe. Even though considerable people get vaccinated, it is not under the control and evolving with new variants. As it is a contagious disease, it is crucial to identify the infected patients as soon as possible. Delays in the identification of the patients or misinterpretation of the results will even worse the situation. Polymerase chain reaction (PCR) test is widely used in the identification of the infected people. Rapid antigen tests also used in this context with very low accuracy. Literature shows that these methods have considerable drawbacks and their reliability depends on so many factors [1, 2].
There are studies in the literature which use machine learning algorithms in the identification of SARS-CoV-2 patients with significant accuracy. These studies vary in many directions including classification between COVID-19 positive and negative cases [3,4,5], separating COVID-19 cough from normal cough [6], COVID-19 detection, prognosis and diagnosis [7,8,9,10,11,12], whether a person is having the risk of COVID-19 or not [13] and differentiating SARS-CoV-2 from other viruses [14,15,16,17]. These studies used different types of data as the input for their model and different set of machine learning algorithms were utilized in these predictions.
Type of the input data used in these models plays a crucial role in the accuracy of the prediction despite the machine learning algorithms. In the literature, varieties of data were used such as genome sequences [15], transcriptome data [16], recorded voices [6], symptoms, clinical and morphological features of the patients [3,4,5, 7,8,9, 12, 13, 17] and X-ray images [10].
Studying the literature shows that molecular data are rarely used in these machine learning related diagnosis of SARS-CoV-2. However, transcriptome data are the most widely used data in the investigation of diseases in molecular level [18]. Further, previous biological studies showed that transcriptome profiling gives a better understanding of COVID-19 pathogenesis in SARS-Cov2 patients [19, 20]. Previous studies also showed the connection between transcriptome data and COVID-19 severity [21]. This study utilizes these findings in order to diagnose the SARS-CoV2 patients using their transcriptome data and different machine learning algorithms.
Altogether seven different classification algorithms are used along with different feature selection techniques. Differently expressed genes (DEGs) between COVID-19 patients and non-COVID individuals also studied here. Gene ontology (GO) analysis on the DEGs shows that they are mostly related to immune, inflammatory and defense response activities. Using the selected features for this stratification shows that features selected using mutual information (or DEGs) along with naïve Bayes (or SVM) classifier gives the best accuracy (0.98 ± 0.04) among all the studied models.
Materials and Methods
Materials
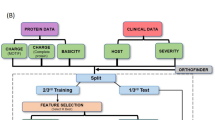
Publicly available high-throughput sequencing transcriptome data from GEO omnibus with the accession number of GSE 189199 is used in this study. They used the pulmonary draining lymph nodes collected at autopsy from 22 lethal COVID-19 cases and 28 control samples. Control lymph nodes were collected from a range of histomorphological sequelae.
Feature Selection Methods
Forward Feature Selection
Forward feature selection is a greedy algorithm starts with an empty set of selected features. This algorithm iteratively finds the best combination of features of the model. The best feature of the prediction will be added to the empty set at the first iteration and then the second one will be selected and added to the existing feature. This process will continue either the defined number of features are selected or the performance of the model remains unchanged.
Feature Importance
In this method, a score called feature importance is calculated for all the input features. This score gives the importance of a particular feature for the given problem. Features with high score are considered as the important features of that problem. It checks whether the means of the samples are from same distribution or not, hence the variance between groups. It can be measured in many ways and those methods can be grouped under two main groups such as model agnostic methods and model-dependent methods. Here, model-dependent methods are specific to a particular model. However, they can be used as separated methods as well.
On the contrary, the other method uses a variety of criteria including correlation criteria, single value prediction and permutation feature importance to calculate the feature importance. First criteria uses any correlation measures to simply correlate the features with target value and calculate the feature importance score. Second one uses every single feature as the input to the model and calculates the importance of that feature. Final one uses an idea like observing the model prediction when there is a change in the value of a single variable. This is done by applying permutations to the algorithm.
Mutual Information
Non-linear relationship between two variables are measured in mutual information. Further, this measure shows the quantity of the information which can be obtained about a random variable by using another. High mutual information refers that those two variables are closely connected and there is a large reduction of uncertainty. For two independent random variables, this value should be zero.
Mutual information can be expressed as,
Here, H(X) and H(Y) are the marginal entropies, H(X|Y) and H(Y|X) are the conditional entropies and H(X,Y) is the joint entropy of X and Y.
Machine Learning Algorithms
Decision Tree Classifier
This is a supervised machine learning algorithm where the data are continuously split based on feature values. In every step, there is a question on one selected feature from the data and the whole data will be split into two based on the answer of that question. This process can be viewed as a binary tree, where the tree is built via a process called as binary recursive partitioning. This process continues until we met one target value.
Random Forest Classifier
Random forest is an ensemble algorithm where more than one algorithms are combined for classifying objects. Here, in random forest, multiple decision trees are applied on randomly selected subset of training data. Votes from all those decision trees are then aggregated to predict the final output.
Naïve Bayes Classifier
This is a probabilistic classifier based on Bayes theorem. This algorithm works under the assumption of all the features are independent. Here, the given feature values will be used to calculate the probability of each class to assign the new instance. The new instance will be assigned to the class with the highest probability.
Support Vector Machines (SVM)
SVMs are powerful classification algorithms under the supervised learning. Here, dataset is divided into classes to find a maximum marginal hyper plane. Hence, the distance between this hyper plane and the closest data points of each class from that hyper plane (support vectors) is maximized. This is an iterative process to generate the hyper plane to separate the data and finally it will choose the proper hyper plane.
K-Nearest Neighbor (KNN) Classifier
This is a supervised classification algorithm based on a number of nearest neighbors. This algorithm works based on the similarity between features. The new data point will be assigned to a class with high number of closely matched data points. Number of closest data points to be considered (K) can be defined by the user. For each data point in the training set, the distance should be calculated from the new data. Based on the distance value, K closest point will be chosen and the new data point will be assigned to the class with the maximum number of closest data points.
Perceptron
Perceptron is another supervised algorithm for binary classification. This is the possible simplest artificial neural network. This will take the number of input features and produce one binary output. Different weights will be calculated for each features during the training phase and used on the test data. Calculated value in the testing phase will be checked against a threshold value. The output depends on this threshold. If it is greater than the threshold output is 1 (or 0) and else it is 0 (or 1).
Cross-validation
Fivefold Cross-validation
Cross-validation is a powerful tool which gives us a confident on the performance of our model. Because of the limited number of samples, fivefold cross-validation is used here. In this method, entire data is randomly divided into five groups. In each iteration, one group is used for testing and other four will be used in the training. This process will be repeated for five times and in each step the group used for testing will change.
Leave One Out Cross-validation (LOOCV)
This is another cross-validation technique used in the validation of our model. Here, a single data will be used as the test data in each iteration and all the others in the training of the model. This is also exercised here because of the number of samples. This will further validate our result by fivefold cross-validation.
Results
Feature Selection
As the data value in a wide range, before the feature selection and model building data are normalized between 0 and 1. The normalized value is used in the study.
Forward Feature Selection
Using forward feature selection, algorithm shows that top three features can do this classification almost perfectly. Using ITGB2, ATF6 and ARHGEF1 gives the best accuracy and adding more features does not change the accuracy. Gene Ontology (GO) analysis [22] on the selected genes shows that these genes are highly related to integrin alphaL-beta2 complex (p value = 0.12), which is connected to immune response-related activities [23].
As there is a performance drop after eight features, initially eight features are selected and used from this method and compared with other feature selection methods Fig. 1a. However, supplementary figure shows that even using those top three features does not compete with other feature selection methods.

Selected features using different algorithms along with their values. a Top ten features selected using forward feature selection algorithm with their accuracy value on the model, b top 25 features selected using feature importance is plotted against their feature importance values and c mutual information selected top 25 features are with their mutual information value
Feature Importance
Feature importance of top 25 features are represented in Fig. 1c. These are the features initially selected to be used in the classification model. GO analysis [22] on these studies shows that they are related to immune system process (p value = 0.007), regulation of response to stimulus (0.015), platelet degranulation (p value = 0.018), positive regulation of response to stimulus (p value = 0.03) and amyloid fibril formation (p value = 0.04).
Mutual Information
Again, top 25 features are selected using mutual information values and used in the classification. Checking the ontology terms of those genes shows that they are highly related to immune-related activities such as immune system process, immune response, cell activation, hemopoiesis and regulation of immune system process with very low p values (up to 10−6).
List of all the selected features and the complete list of related GO terms (highest with low p values are presented here) can be found in the supplementary file.
Differently Expressed Genes (DEGs)
Differently expressed genes are studied between SARS-CoV-2 cases and the control samples using TCC:GUI [24]. It shows that totally there are 1283 differently expressed genes between COVID-19 patients and control (Fig. 2). GO analysis on these DEGs gives prominent terms related to immune and defense response. It includes immune response, defense response, response to stress, inflammatory response and the whole list is found with very low p value (supplementary material).

General studies on the measured transcriptome. COVID-19 samples are compared with non-COVID. a Heat map of the differently expressed genes between COVID-19 and control, b 3D PCA visualization between COVID-19 and control, c volcano plot of the genes. Right—up-regulated genes and left—down-regulated genes d MA plot of genes. Differently expressed genes are represented in red
Machine Learning Algorithms
Seven different machine learning models are used here with four different set of selected features. Fivefold cross-validation is used in the validation of the models. Accuracy is used in the accuracy measure of the models.
Random Forest Classifier
Initially, 25 features from feature importance, mutual information and DEGs, and 8 features from forward feature selection are individually used in the classification using random forest (Fig. 3). In this case, mutual information gives best accuracy (0.96 ± 0.09) (Table 1) and all these results are after fivefold cross-validation. This prediction accuracy is validated with precision of 1.0 and recall value of 0.91, where the false negative prediction is one patient out of 20, 0.05% (Fig. 4a).

Accuracies by different machine leaning algorithms in the prediction of COVID-19 patients after fivefold cross-validation. a Feature importance selected features are used with different classification algorithms. b Mutual information selected features c DEGs are used d Features selected using forward feature selection algorithm

Confusion matrix of the best prediction form each classifier: a top 25 features from mutual information using random forest b mutual information selected top 25 features in the diagnosis using naïve Bayes c twenty five DEGs in the diagnosis using linear SVM d Decision tree e KNN f perceptron
Naïve Bayes Classifier
Same number of features are used in this model as well. Here too, mutual information selected features give the best accuracy (0.98 ± 0.04), which is the best accuracy among all the models (Table 1). This result is confirmed using LOOCV (0.96 ± 0.2) as well. False negative of this study shows that, all the 20 patients used in testing are correctly predicted with 0% false negative (Fig. 4b). Precision and recall evaluation shows both of them are 1.0.
Support Vector Machines
Here, linear and polynomial SVMs are used with all those features. In linear SVM, DEGs give the best accuracy (0.98 ± 0.04), best accuracy of the study (Table 1). Table 1 shows that this is the best model using LOOCV as well. Feature importance selected features gives the best accuracy in SVM-polynomial model (0.96 ± 0.09). Degree of the polynomial SVM is set to 3 in this study. Here also, there are no false negatives (Fig. 4c) in the diagnosis, which yields precision and recall of 1.
Decision Tree Classifier
In overall, this is the worst performing model with the highest accuracy of 0.88 ± 0.1. Feature importance selected features and forward feature selection features give this accuracy. Figure 4d shows that out of 20 patients, 1 false negative and 3 false positives, which is the reason for this low accuracy. Study the precision and recall also shows comparably low values (0.75 and 0.9, respectively).
K-Nearest Neighbor Classifier
Even though decision tree performs worst on the whole, KNN-classifier gives the worst accuracy (0.78 ± 0.1), while using forward feature selection features. Best performance of this model is gained using feature importance features (0.96 ± 0.09). Here, two nearest neighbors are used (k = 2). Even though there are no false positives in this method, one false positive id identified with precision of 0.89 and recall of 1.
Perceptron
In the perceptron model, feature importance selected features give the best accuracy (0.94 ± 0.09). Here also, one false positive is identified without any false positive with precision of 0.91 and recall of 1.
Discussion
Three different feature selection methods are used in this study to select suitable features. Twenty five features are selected using feature importance and mutual information. In forward feature selection method, it started with ten features (in the parameter setting). However, after three features, there is no improvement in the accuracy of the model. This shows that those three features can perform better than other features in this prediction.
Initially, the model is built using these features and used as the main results. Then, first three features from all the methods are used in the same way and presented in the supplementary material. It shows that there is a clear improvement in the accuracies of forward feature selected features (three features) compared to ten features. However, it is less than the highest accuracy of this study.
Comparing the accuracies shows that feature importance selected features performs better on the whole with all the models. Anyhow, the best accuracy is obtained by mutual information selected 25 features with naïve Bayes classifier. The same accuracy is gained by 25 DEGs along with SVM (linear and polynomial).
Comparing this accuracy with previous studies shows that transcriptome data performs better in this classification compared to non-molecular data. Using routine blood samples in the same study gave the accuracy value between 82 and 86% [3]. Laboratory, clinical and demographic data also used in this study and achieved the average area under the curve (AUC) value of 0.92 [7]. Another study used number of lymphocytes, leukocytes and eosinophils in the COVID-19 diagnosis and achieved the AUC value of 0.85 [8]. Eight general binary features including sex, known contact with infected people and appearance of clinical symptoms are used in the classification and reported the AUC value greater than 90% [11]. Using symptoms and comorbidities details of the patients along with their general information provided the highest accuracy value of 94.3% [4] and 92% [13] in the same prediction.
Even using transcriptome data in the same diagnosis gave the highest accuracy value of 0.938 using support vector machines [25]. Their work is almost similar to this work, where the number of features used is different. Their highest accuracy is with 168 features selected using Boruta feature filtering. However, using transcriptome data gave the accuracy value of 0.98 along with multi-layer perceptron in another study [26]. However, the drawback of their study is they did feature extraction, not the feature selection. Hence, using this study it is impossible to find out the genes related to the diagnosis of COVID-19. Also, the variance of their accuracy is not mentioned in their study. If it is high, the significance of their accuracy will be low compared to this study. Another study used miRNAs in the same prediction and diagnosis using deep neural networks and achieved the maximum area under the ROC curve value of 0.79 and F1 value of 0.74 [27].
False negative cases are the worst problem in this case, where they can spread the virus without their knowledge. This study analyses the false negatives of the prediction while using 60% of the data for training and 40% for testing. Here, this study shows the maximum false negative case of 1% and minimum of 0%, where literature shows the higher risk of false negative in PCR test. In PCR test, the initial false negative rate can be up to 54% [28]. In some cases, the presence of COVID-19 was identified after fifth PCR test in an admitted patient [29].
Comparing this study with the existing studies shows that this study utilizes the power of feature selection methods and gain the maximum accuracy with low variance. Here, more than one feature selection methods are used and the performances are compared and the maximum is selected. Similar existing studies using more features comparing to this study showed a lower performance than this study. Further, this study presents the set of identified features which can be used in further biologically related studies or analyses. Also, it validated the accuracy using cross-validation. This step is very important, because high variance may lead to an insignificant result.
Conclusion
This study uses high-throughput sequencing transcriptome data in the classification of COVID-19 patients against non-COVID. Feature importance, mutual information and forward feature selection algorithms are used in the feature selection. Differently expressed genes also studied here between SARS-CoV-2 and control samples. GO analysis on the selected features and DEGs shows a very close relationship with immune and inflammatory responses. Those selected features (25 features from feature importance, mutual information and DEGs and eight (or three) features selected from forward feature selection) are used with seven different classification algorithms. This study shows that mutual information selected features along with naïve Bayes classifier or DEGs with SVM give the best accuracy value (0.98 ± 0.04) in this classification. Further, it shows that molecular data can give more accurate prediction of COVID-19 against non-COVID-19 compared to other data.
Data availability
Publicly available high-throughput sequencing transcriptome data from GEO omnibus with the accession number of GSE 189199 is used in this study.
References
Axell-House D, Lavingia R, Rafferty M, Clark E, Amirian E, Chiao E. The estimation of diagnostic accuracy of tests for COVID-19: a scoping review. J Infect. 2020;81(5):681–97.
Ferté T, Ramel V, Cazanave C, et al. Accuracy of COVID-19 rapid antigenic tests compared to RT-PCR in a student population: the StudyCov study. J Clin Virol. 2021;141:104878.
Brinati D, Campagner A, Ferrari D, Locatelli M, Banfi G, Cabitza F. Detection of COVID-19 infection from routine blood exams with machine learning: a feasibility study. J Med Syst. 2020. https://doi.org/10.1007/s10916-020-01597-4.
Muhammad LJ, Algehyne EA, Usman SS, Ahmad A, Chakraborty C. Supervised machine learning models for prediction of COVID-19 infection using epidemiology dataset. SN Comput Sci. 2021;2:11.
Savla H, Mehta V, Mangrulkar R. Prediction and diagnosis of COVID-19 using machine learning algorithms. Int J Recent Technol Eng (IJRTE). 2020;9(3):678–83.
Madhurananda P, Marisa K, Robin W, Thomas N. COVID-19 cough classification using machine learning and global smartphone recordings. Comput Biol Med. 2021;135:104572.
Fernandes F, Oliveira Td, Teixeira C, Batista A, Costa GD, Filho AC. A multipurpose machine learning approach to predict COVID-19 negative prognosis in São Paulo Brazil. Sci Rep. 2021;11(1):3343.
Batista dM, Filipe A, Luiz MJ, Henrique RDT, Filho PC, Dias A. COVID-19 diagnosis prediction in emergency care patients: a machine learning approach. medrxiv. 2020.
Matta D, Saraf M. Prediction of COVID-19 using Machine Learning Techniques [Dissertation]. http://urn.kb.se/resolve?urn=urn:nbn:se:bth-20232. 2020.
Salam MA, Taha S, Ramadan M. COVID-19 detection using federated machine learning. PLoS ONE. 2021;16(6):e0252573.
Zoabi Y, Deri-Rozov S, Shomron N. Machine learning-based prediction of COVID-19 diagnosis based on symptoms. npj Digital Medicine. 2021. https://doi.org/10.1038/s41746-020-00372-6.
Assaf D, Gutman Y, Neuman Y, et al. Utilization of machine-learning models to accurately predict the risk for critical COVID-19. Intern Emerg Med. 2020;15(8):1435–43.
Annwesha BM, Somsubhra G, Dharmpal S, Sourav M. An intelligent system for prediction of COVID-19 case using machine learning framework-logistic regression. J Phys: Conf Ser. 2021;1797: 012011.
Ng D, Granados A, Santos Y, et al. A diagnostic host response biosignature for COVID-19 from RNA profiling of nasal swabs and blood. Sci Adv. 2021. https://doi.org/10.1126/sciadv.abe5984.
Arslan H. Machine learning methods for COVID-19 prediction using human genomic data. Proceedings. 2021;74(1):20.
Zhang Y, Li H, Zeng T, et al. Identifying transcriptomic signatures and rules for SARS-CoV-2 infection. Front Cell Dev Biol. 2021;8:627302.
Li W, Ma J, Shende N, et al. Using machine learning of clinical data to diagnose COVID-19: a systematic review and meta-analysis. BMC Med Inform Decis Mak. 2020;20(1):1–3.
Casamassimi A, Federico A, Rienzo M, Esposito S, Ciccodicola A. Transcriptome profiling in human diseases: new advances and perspectives. Int J Mol Sci. 2017;18(8):1652.
Andrea RD, Prathyusha B, Katherine AO, et al. Comprehensive transcriptomic analysis of COVID-19 blood, lung, and airway. Sci Rep. 2021;11(1):1–19.
Chakraborty C, Sharma A, Bhattacharya M, Zayed H, Lee S. Understanding gene expression and transcriptome profiling of COVID-19: an initiative towards the mapping of protective immunity genes against SARS-CoV-2 infection. Front Immunol. 2021;12:724936.
Jain R, Ramaswamy S, Harilal D, et al. Host transcriptomic profiling of COVID-19 patients with mild, moderate, and severe clinical outcomes. Comput Struct Biotechnol J. 2020;19:153–60.
Sebastian B, Steffen G, Martin V, Peter R. Ontologizer 2.0—a multifunctional tool for GO term enrichment analysis and data exploration. Bioinformatics. 2008;24:1650–1.
Ashok D, Anjana C, George H, Shankar S. Integrin beta-2. UCSD Molecule Pages. 2013;2:33–47.
Wei S, Jianqiang S, Kentaro S, Koji K. TCC-GUI: a Shiny-based application for differential expression analysis of RNA-Seq count data. BMC Res Notes. 2019;12(1):1–6.
Chen L, Li Z, Zeng T, et al. Identifying COVID-19-specific transcriptomic biomarkers with machine learning methods. BioMed Research International. 2021.
Liu X, Hasan M, Ahmed K. Machine learning to analyse omic-data for COVID-19 diagnosis and prognosis. BMC Bioinformatics. 2023. https://doi.org/10.1186/s12859-022-05127-6.
Bugnon L, Raad J, Merino G, et al. Deep Learning for the discovery of new pre-miRNAs: helping the fight against COVID-19. Machine Learning with Applications. 2021;6:100150.
Arevalo-Rodriguez I, Buitrago-Garcia D, Simancas-Racines D, et al. False-negative results of initial RT-PCR assays for COVID-19: a systematic review. PLoS ONE. 2020;15(12):e0242958.
Feng H, Liu Y, Lv M, Zhong J. A case report of COVID-19 with false negative RT-PCR test: necessity of chest CT. Jpn J Radiol. 2020;38(5):409–10.
Funding
The author received no funding for this work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares there are no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the topical collection “Computer Aided Methods to Combat COVID-19 Pandemic” guest edited by David Clifton, Matthew Brown, Yuan-Ting Zhang and Tapabrata Chakraborty.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Jeyananthan, P. SARS-CoV-2 Diagnosis Using Transcriptome Data: A Machine Learning Approach. SN COMPUT. SCI. 4, 218 (2023). https://doi.org/10.1007/s42979-023-01703-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42979-023-01703-6