
Abstract
Background
Managed Access Agreements (MAAs) are a commercial arrangement that provide patients earlier access to innovative health technologies while uncertainties in the evidence base are resolved through data collection. In the UK, data collection agreements (DCAs) outline the evidence that will be collected during the MAA period and are intended to resolve uncertainties in the clinical- and cost-effectiveness of a technology sufficient for the National Institute of Health and Care Excellence (NICE) committee to make a final decision on reimbursement.
Objective
The aim of this study was to identify the primary uncertainties leading to a recommendation for entry to the Cancer Drugs Fund (CDF) and evaluate how the corresponding DCAs attempt to address these.
Methods
A database of MAAs agreed within the CDF was compiled with coverage between July 2016 and December 2020 (the time during which evidence generation was routinely collected within the CDF up until the time of analysis). Uncertainties in the evidence base for technologies entering the CDF were analysed alongside the outcomes planned for data collection during the MAA. These data provide an overview of the key uncertainties surrounding health technologies in the CDF on entry and the types of evidence targeted by DCAs.
Results
In the assessment of 39 Cancer Drugs Fund (CDF) cases, NICE committees identified a total of 108 key uncertainties in cost-effectiveness estimates. Overall survival was the most commonly identified uncertainty, followed by generalisability of the evidence to the target population. DCAs specified a range of outcomes relevant to understanding the clinical effectiveness of the technology, though fewer than half (43.6%) of the DCAs addressed all the key uncertainties identified by the NICE committee.
Conclusion
The analysis indicated that data collection within the CDF is not sufficient to resolve all the uncertainties identified by the NICE committee, meaning that other approaches will be needed at re-appraisal to ensure that the NICE committee can reach a final decision on reimbursement.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Across 39 technologies that entered into the Cancer Drugs Fund, 108 uncertainties were identified. The most common uncertainties included overall survival estimates, as well as the generalisability of the evidence to the target population. |
Fewer than half of the corresponding data collection agreements addressed all the key uncertainties identified by committees. Analysis indicated that the data collection is therefore not sufficient to resolve all feasible uncertainties, meaning the data collection agreements could be made more comprehensive to attempt to resolve more of the uncertainties. |
1 Introduction
Managed access agreements (MAAs) are commercial arrangements between payers and industry that provide patient access to health technologies when further evidence is required to establish clinical effectiveness and cost-effectiveness. Finding the optimum balance between access and ensuring that technologies deliver value to patients and the health service is a significant challenge for policy-makers [1]. In the UK, MAAs are used to provide temporary access to promising technologies in areas of high unmet need, and a data collection agreement (DCA) is developed in collaboration with the manufacturer that aims to inform a final decision on routine commissioning at the end of the MAA. Historically, the majority of MAAs in the UK are targeted towards cancer treatments. This is largely due to the establishment of the Cancer Drugs Fund (CDF) [2, 3] originally set up in 2011 and reformed in 2016 to enable access to high-cost cancer treatments with highly uncertain clinical benefits. The use of MAAs in the UK is now set to expand following the introduction of the Innovative Medicines Fund (IMF) [4], which will provide equivalent funding for MAAs with technologies in non-cancer indications.
MAAs largely include ‘innovative’ technologies – technologies that address a high unmet need, offer a step-change in treatment and present challenges for evidence generation [5]. They typically span 3–5 years, during which forthcoming trial and real-world evidence generated in NHS settings are used to inform decision-making by a NICE committee at the end of the agreement. A growing number of MAAs have reached completion, and the majority have led to a positive recommendation; as of Dec 2020, more than 53 MAAs have been funded through the CDF. Of these, 26 have been reviewed following data collection, 22 (84%) of which have been recommended for routine commissioning. From the first 24 technologies, routine commissioning was even higher, with an acceptance of 87.5% at re-appraisal [6]. This suggests that data collected during the MAA has been sufficient to aid decision-making by NICE. However, appraisals of MAAs have found that uncertainties may not be resolved during the agreement period, and final decisions on commissioning remain complicated [7].
The 2022 NICE manual states explicit consideration of the uncertainties, including the types of uncertainty (parameter, structural etc), the impact on cost-effective estimates and highlighting those that are unlikely to be resolved with additional evidence generation or expert input [8]. Where uncertainties persist at the end of the MAA period, a commercial agreement between the payer and the company may compensate for the remaining uncertainty and balance the risks associated to enable continued patient access. For example, companies offering a higher discount upon CDF exit, which may be commercially challenging, especially for technologies used for multiple indications within the NHS.
From both the company and reimbursement agency perspectives, it would be preferable if MAAs contained clearly defined DCAs appropriate to resolve specific uncertainties in the evidence [9]. However, the DCAs may not always be able to resolve the highlighted uncertainties. Trial data and additional follow-up are important, but are often not able to solve all immaturity issues. Additionally, the Systematic Anti-Cancer Therapy (SACT) dataset, a primary database for oncology treatment data, may not be regarded as robust enough to use in an economic evaluation [6].
1.1 Aims
This study aims to examine the evidential uncertainties for technologies entering the CDF, and how DCAs attempted to address these. A previous analysis highlighted the largest areas of uncertainty for technologies entering MAAs related to clinical effectiveness (88%) and generalisability (50%) [10]. An updated review is important to highlight the most common areas of the uncertainties that remain in a now more established managed access program. This will aid companies in appraisal by highlighting uncertainties commonly identified, to ensure adequate data are available prior to submission. The analysis compared uncertainties identified by NICE and the corresponding DCAs to assess how thoroughly uncertainties were addressed to enable the MAAs to better achieve their objectives. Accordingly, this study sought to answer the following research questions:
-
What areas of uncertainty are identified by NICE for technologies recommended with managed access in the CDF?
-
What data are specified for collection in CDF DCAs?
-
Do DCAs fully address the uncertainties raised by the NICE committee?
2 Methods
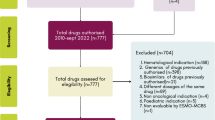
A database was compiled containing information about all technologies that have entered the CDF between 31 July 2016 (following the reforms to the CDF) and 31 December 2020, when this review was carried out. Data collected included information about the uncertainties highlighted by the NICE committee during the HTA process, and the outcomes specified for collection within the DCAs.
2.1 Data Collection Sources
Details of current CDF technologies were extracted, including the key uncertainties highlighted by the committee, the indication and licensing information, characteristics of the target population, overall survival data and characteristics of the evidence base at the time of appraisal. Data were derived from publicly available documents identified from the NICE website [11] including NICE committee papers, the final appraisal documents (FAD) and the DCAs. Where required, additional information about the technologies was identified from EMA and MHRA licensing documentation [12, 13]. Where multiple committee meetings took place, uncertainties identified by the committee in the final committee meeting prior to the MAA were selected, as these remain once all evidence was presented. DCAs specified outcomes that would be measured within new or ongoing clinical trials, as well as outcomes to be measured using real-world data sources. Where data were redacted due to the confidentiality of data sources or commercial negotiations, there data points were recorded as not accessible. This affected a minority of data, most commonly the expected data cuts for clinical trials.
2.2 Data Extraction
The database was piloted on 15% of MAAs, and further refined before use. Data were extracted by a single reviewer (LT or SH), and 100% of records were reviewed for accuracy by a senior reviewer. Data were extracted according to the source description, with no or limited subjective interpretation on behalf of the research team. The data extraction table was shared with the managed access team at NICE, and edits were made to data points where further clarity was available.
2.3 Data Analysis
The data were analysed using a mixed methods approach. Uncertainties were defined as meaningful uncertainties raised by the NICE committee in its final guidance, often found in Sect. 4 of the MAA. The NICE committee highlights those uncertainties in the appraisal that it considers to plausibly have a meaningful influence on cost-effectiveness estimates, and which therefore influenced their final recommendation. Extracted uncertainties were derived from specific statements of uncertainty (e.g. short-term trial data on overall survival) and grouped inductively and hierarchically using thematic network analysis [14]. Uncertainties were first considered with the intention of identifying overall themes. These themes were refined on the basis of a process of cross-comparison, to lead to the formation of the key questions that best represented the overall themes of uncertainties identified. Uncertainties were then coded according to the overall theme which they addressed and to identify sub-themes. For example, an uncertainty that pertained to question 1 (How well does this treatment work, and for whom?) may also be coded as relating to uncertainty in overall survival estimates and the long-term durability of the treatment effect.
Identified data collection areas were extracted from the DCA and classified using the analysis of uncertainties as an analysis frame. Classification of data collection outcomes was conducted solely on the basis of the DCA, and blind to the uncertainties raised by the NICE committee for that appraisal.
When drawing comparisons between uncertainties identified by NICE and the planned data collection, an uncertainty was considered to be addressed in the DCA when it was stated in Sect. 3 of the DCA: areas of clinical uncertainty to be addressed by the agreement.
3 Results
A total of 39 MAAs funded by the CDF were identified within the analytical period (31 July 2016–31 December 2020). Some cancer drugs entered more than one MAA (i.e. across multiple indications, e.g. pembrolizumab, nivolumab, ateolizumab, olaparib, niraparib and rucaparib).
3.1 Uncertainties Identified
In the assessment of 39 Cancer Drugs Fund (CDF) cases, NICE committees identified a total of 108 key uncertainties in cost-effectiveness estimates. On average, each appraisal had three identified uncertainties, though this varied across appraisals: specifically, 6 out of 39 (15.4%) cases had just one key uncertainty, 7 out of 39 (17.9%) had two key uncertainties, 16 out of 39 (41.0%) had three key uncertainties and 10 out of 39 (25.6%) had four or more key uncertainties. Uncertainties were categorised into five key questions:
-
(1)
How well does this treatment work, and for whom?
-
(2)
How well does this evidence generalise to the UK?
-
(3)
How do we evaluate the clinical evidence for this treatment?
-
(4)
How do we value the benefits of this treatment?
-
(5)
How long is treatment used?
The key questions and subgroups within these can be seen in Table 1. The most prevalent uncertainty across CDF appraisals was long-term overall survival (OS), commonly due to the immaturity of the data presented during the appraisal, and the lifetime horizon of many cancer indications leading to uncertain extrapolation (occurring in 34 out of 48, or 70.8%, of OS uncertainties). In some cases (6 out of 48, or 12.5%) the uncertainty related to specific comparators, or heterogeneity in estimates of OS. Progression-free survival (PFS) was also a common uncertainty due to the immaturity of clinical data. In two technology appraisals (TAs), the committees also highlighted uncertainties in how PFS related to OS, for example, due to converging or diverging curves that challenged clinical interpretation. Uncertainties surrounding the safety of technologies were not relatively common (2%) and, where these occurred, were related to immunotherapies.
Nearly a quarter of uncertainties (23.1%) were related to the generalisability of the evidence to the UK context, which were related to variability in model assumptions to NHS treatment pathways (17 out of 108, or 15.7%), both concomitant and subsequent to the technology under evaluation, and the use of treatment-stopping rules in NHS practice. These also related to the uncertainty surrounding baseline risk and previous treatments received.
Uncertainty concerning the clinical benefit of the technology within the follow-up period of the evidence accounted for only 7.4% of the uncertainties, and was related to the use of network meta-analysis and the direct limitations from trials (for example, risk of bias issues in non-randomised or single-arm trials). The uncertainty around the valuation of the benefits was attributable to uncertainties around health-related quality of life estimates (HRQoL) used in economic models. Finally, the treatment duration was an area of uncertainty with specific relation to the expected time on treatment.
3.2 Data Collection Agreements
There were 166 data points planned for collection within DCAs for the 39 CDF MAAs, with the majority specifying between 1 and 4 outcomes to be collected. A significant minority, 44 out of 166 (26.50%), of these data points were within DCAs for just two MAAs, both for non-histology-dependent tumours (TA630 larotrectinib and TA644 entrectinib). Data collection was typically based on ongoing or new clinical trials, though most DCAs also included data collection from NHS registries including the Systemic Anti-Cancer Therapies (SACT) and NHS England CDF prior approval (Blueteq) databases.
The data collection outcomes were categorised into one of seven themes (shown in Table 2):
-
o
Survival.
-
o
Patient characteristics.
-
o
Treatment pathway.
-
o
Health-related quality of life.
-
o
Testing.
-
o
Efficacy.
-
o
Real-world use.
Data relating to the treatment pathway accounted for 22.29% of the data within DCAs and were present in a third of all CDF DCAs. These related to the prior or subsequent treatments, and the way in which start and stopping criteria would be used in practice. All of the above would typically affect the cost-effectiveness of a technology and are amenable to data collection within NHS settings. Prior and subsequent treatment outcomes included the type of therapy given, the number of patients/applications to start treatment (in one case categorised by tumour site), treatments given to relapsing patients and the number of previous lines of therapy. Data related to start and stopping criteria included the time to treatment discontinuation, the number of patients starting treatment within the timeframe of the MAA and, in one case, the numbers of patients who are still on the treatment after 2 years with residual disease.
Consistent with being the most common uncertainty across appraisals (listed in 36 out of 39 DCAs), OS data accounted for 21.69% of data collections points across DCAs. OS data were typically requested to augment the immaturity of the available evidence in relation to the lifetime horizon of the indication; however, as MAAs only last a short number of years, their usefulness for generating OS data is limited to comparisons against survival curves from RCTs and the assumptions that underpin extrapolations. Real-world data for progression-free survival included outcomes on 5-year progression, treatment-free intervals and progression of disease.
Patient characteristics, specifically the generalisability of the trial population to the target NHS population, accounted for 12.65% of the uncertainties specified, and most frequently referred to baseline characteristics, with some DCAs specifically requiring information about the age and gender of patients who receive treatment. The area of uncertainty that these additional data seek to resolve referred most commonly to the generalisability of the patient population to the intended use population. These data were required where demographic characteristics were of prognostic value for predicting clinical treatment outcomes, and/or for predicting costs incurred across the time horizon of treatment. One DCA specified outcomes on the distribution of tumour sites in a real-world setting.
Data points related to treatment efficacy included both clinical and safety outcomes. Surprisingly however, data for health-related quality of life accounted for just 3.01% of the outcomes specified in DCAs.
Real-world use of the drug accounted for 18.67% of the data collection points. Information on the treatment duration and comparator treatments, including the content and outcome of best supportive care, were included as well as the real-world response and progression of patients.
The frequency of outcomes collected across the 39 MAAs is shown in Fig. 1.

Outcomes to be collected across CDF MAAs. BSC best supportive care, HRQoL health-related quality of life, MAA managed access agreement
3.3 Relationship Between the Uncertainties and Data Collection Agreements
A comparison between uncertainties raised by the NICE committee and DCAs is shown in Fig. 2. In six DCAs (6 out of 39, or 15.39%), data to be collected perfectly matched the uncertainties raised. However, less than half (17 out of 39, or 43.59%) of the DCAs incorporated data points to address all the uncertainties. Areas of uncertainty commonly not resolved included the generalisability of the treatment, treatment duration and efficacy-related uncertainties. Health-related quality of life had no unresolved uncertainties, and two TAs included data collection on HRQoL when it wasn’t highlighted by the committee. In all, 1 out of 22 (TA446 Brentuximab Vedotin; 4.55%) of the TAs that had remaining areas of uncertainty did not address any uncertainty highlighted in appraisal. However, this was the first topic that entered the CDF, and therefore is likely not an accurate representation of the current managed access procedures. The uncertainty that was highlighted concerned the proportion of treated patients, whereas the data collection agreement collected data on whether the patient had had a stem cell transplant before or after the treatment, and best supportive care. In addition to the uncertainties raised by the committee, 30 out of 39 (76.92%) DCAs outlined additional data collection that was not highlighted as an uncertainty. Common additional areas of data collection included the treatment timings, baseline characteristics of patients and progression-free survival data.

The level at which DCAs addressed the uncertainties raised by the NICE committees. Data was categorised into the following: data collection plans addressed all identified uncertainties, data collection plans addressed some identified uncertainties, and data collection plans including additional data collection not specifically raised by NICE committees. CDF cancer drugs fund, DCA data collection agreement, MAA managed access agreement
4 Discussion
The stated purpose of the CDF is to support access to innovative cancer technologies while generating evidence sufficient to resolve uncertainties in the clinical effectiveness and cost-effectiveness that would enable a decision on routine commissioning. This review set out to analyse the areas of uncertainty in cost-effectiveness estimates identified by the NICE HTA committees that led to a recommendation for MA in the CDF and relate these to the formation of DCAs within MAAs. This analysis suggests that methods within published DCAs only partially address the uncertainties raised by NICEs committees prior to entering the CDF: under half the technologies entering the CDF were accompanied with a DCA fully addressing the uncertainties raised by the committee. Ultimately, this necessitates NICE committees during re-appraisal to make final determinations regarding routine commissioning amid persistent and substantial uncertainties in cost-effectiveness assessments. Data collection agreements offer a means for continuous technology evaluation; however, even when fully adhered to, they may not definitively resolve initial uncertainties. This can be attributed to various factors, including inadequate data collection durations, model structure and assumptions, limited patient numbers and substantial heterogeneity in collected data, which are all prevalent challenges in oncology. Moreover, the intricate mechanisms of action in oncology drugs lead to diverse outcomes among different patient groups, further contributing to ongoing uncertainties in cost-effectiveness estimates at the conclusion of the MAA period. As a result, a favourable recommendation may pose heightened risk for the health service. Consequently, decision-makers may face the dilemma of either accepting increased risk in their recommendations or exploring commercial arrangements for risk-sharing with manufacturers. This latter option could prove challenging, especially towards the end of the MAA period, as NICE committee chairs have noted manufacturers’ reluctancy to adjust prices to align with uncertainties at the MAA’s conclusion compared with during earlier in HTA appraisal [20].
DCAs are developed on the basis of a series of conversations between representatives from NICE and NHS England CDF clinical leads. In principle, DCAs represent the evidence generation that is considered to be both feasible and meaningful for decision-making at re-appraisal. Within the context of an MAA, it may not be feasible to collect some data points, even if these pertain to meaningful uncertainties in cost-effectiveness estimates. Consistent with an older analysis of health technology assessments (HTAs) [10], our findings showed that OS was the most common uncertainty with technologies entering the CDF, but it is unlikely that a further 3 years of SACT data would resolve this uncertainty for indications not at the end of life [15]. In many cases for technologies entering the CDF, and perhaps increasingly in the context of ongoing improvement in cancer outcomes, meaningful estimates of overall survival may only be measured with longer follow-up than can be captured during the follow-up of phase III trials and the MAA period.
Nevertheless, further data from ongoing RCTs may be useful. The feasibility of data collection and the potential to address uncertainties should be considered prior to a recommendation for managed access to ensure that meaningful uncertainties can be resolved during the data collection period. Value of information (VoI) analysis is one useful approach that could be used to evaluate the feasibility and benefits of data collection efforts during the MAA. VoI analysis quantifies the potential benefits of collecting additional data or conducting further research to make more informed healthcare decisions. Identifying the most costly areas of uncertainty to resolve would help optimise data collection investments and identify the data needed that are most critical for reducing uncertainty. Additionally, utilising VoI analysis enhances the transparency of the decision-making process and could be a tool to standardise acceptable levels of uncertainty on the basis of expected costs of future data collection. This would enable NICE committees to better assess which drugs would most likely benefit from further evidence generation and where to allocate resources for real-world data collection. This approach would also help NICE committees to determine where managed access is not an appropriate avenue, particularly given the substantial time and resources required by payers and companies, and the potential delay to patient access. Novel approaches to generating new evidence are also needed to support managed access, such as formal expert elicitation methods recently recommended within the updated NICE methods guide for HTA [8].
Interestingly, three-quarters of DCAs included data points not clearly related to uncertainties raised by NICE committees as significant for decision-making. These additional data points within DCAs have increased over time, with the average number of data points within DCAs more than doubling since 2020 (from 2.0 to 4.1). Those data related to clinical outcomes may be included in trial protocols and may therefore be required or have further value for companies. Additionally, companies are proactively considering how to supplement clinical trial data with RWD and are interested in having this reflected in their DCAs. However, requests for further data collected within NHS settings required careful consideration. On the one hand, data collection within DCAs is considerably time and resource intensive for NHS staff, and a burden for patients and carers [20]. There is potential for resource misallocation, where time and resources may be diverted away from other research or operational activities that are more effective in reducing uncertainty. On the other hand, additional real-world data collection within DCAs may produce broader value for the NHS, such as informing clinical commissioning, quality audits or care planning – not forgetting the potential to support further evidence generation for NICE evaluations or MAAs in the same disease area. When ICERs were close to, or exceeded, NICE’s willingness-to-pay threshold, single technology appraisals (STAs) with less mature data (for example, less patients experience an event of interest) were likely to be recommended for the CDF (35%), while STAs with mature data were unlikely to be suitable for the CDF (50%) [16]. Cancer drugs with uncertain OS/PFS data are typically those with immature follow-up, while those with more mature data may be more likely to receive a negative recommendation, if companies are unwilling to make a cost-effective commercial offer.
The data in this analysis highlight common gaps in the evidence presented during HTA. One of the most common uncertainties within cancer drugs subsequently entering the CDF was data about the expected treatment pathway in the NHS and the generalisability of evidence to NHS patients. While this evidence may be most applicable to the real-world evidence that can be generated during an MAA, companies seeking to launch products in the UK ought to consider opportunities to generate this evidence as part of their clinical development; this may accelerate decision-making without the need for an MAA. New access pathways for innovative technologies, such as the Innovative Licensing and Access Pathway (ILAP) [17] include support for companies to generate real-world evidence within NHS settings prior to HTA, and may therefore provide earlier insight into the data that might be needed for decisions on routine commissioning. Uncertainties relating to long-term clinical outcomes including OS and PFS may also be better addressed where companies seek to address heterogeneity in estimates and commit research to establishing reliable surrogate measures.
Similar schemes exist internationally; in Australia, managed entry agreements (MEAs) are special pricing arrangements whereby the new technology is listed at a price justified by the availability of evidence and level of uncertainty of this evidence [19]. However, there is no specified timeframe for reappraisals or adjustment of price, meaning that, without the newly generated evidence directly resolving these uncertainties, there is unlikely to be a change in price structure. Though other issues may exist within this scheme (such as the use of surrogate outcomes, which mean that additional data collection may not be comparable to trial data and in general are associated with increased time to patient access [18]), the more considered time frame allows the flexibility for additional data collection to ensure that pivotal uncertainties can be resolved.
4.1 Limitations
There are several limitations to this analysis. First and foremost, the analysis is dependent on the information reported in publicly available documents. There was a high degree of variability across appraisals in the detail reported about uncertainties raised by the committee and the content of DCAs, and the amount of information redacted from public view also varied across appraisal. Where detail was limited, the team interpreted the available information to categorise uncertainties and data points within the analysis. We also assumed that no additional data points than those listed in the DCA would be collected; this may not be correct, and it is feasible that amendments to the DCA were made during the time period of the MAA and not published. While we took steps to increase consistency in extractions, including the way uncertainties were interpreted and categorised, some level of subjectivity remains in the analysis. We attempted to draw conclusions about the way in which DCAs addressed uncertainties raised by the NICE committee on the basis of planned data collection. This assumption does not take into consideration uncertainties that persist despite data collection (for example, data that conflicts with trial evidence), or failures in data collection. Comparatively few MAAs had reached completion during the analysis period, and therefore it is not yet possible to determine precisely to what extent DCAs have been successful in resolving uncertainties raised by the NICE committee. The launch of the Innovative Medicines Fund (IMF) in 2022 has led to changes in the process, for example, the consideration of the eligibility criteria and specific guidance that the DCAs should be feasible and considered to meaningfully reduce uncertainties. This study shows the type of uncertainties that arise in appraisals that committees have decided to recommend to the MAA scheme, though a future evaluation will be able to determine whether the changes to the IMF/CDF have altered this process. Future research may consider the extent to which DCA data collection was successful and resolved key uncertainties. As well as limitations, this analysis has some key strengths. We presented an in-depth analysis that can be impactful, and through the clear methods used, the findings may be developed upon to reduce inefficiencies across the managed access scheme, thus resulting in the reduction inefficiencies across NHS reimbursement programs, whilst promoting earlier access to patients.
4.2 Conclusion
Overall, this analysis identifies the common uncertainties present in the appraisals that ultimately enter the CDF. Key learnings are to what extent data collection within the CDF is able to address these uncertainties, or whether additional or alternative approaches are needed to support a final decision by the NICE committee. As more appraisals exit the CDF, a further analysis of the success of evidence generation will be useful to guide policy-makers.
Development in research has the potential to improve the usefulness of data collection agreements to reduce uncertainty. Whilst it is unlikely that simply requesting more data to be collected from each managed access technology will resolve all uncertainties, more flexibility in how this data is collected may prove to be a more robust solution. For example, overall survival estimates can only be estimated when patients have passed away, and a standardised 3–5 years for additional data collection may not be appropriate for all technologies.
References
Dabbous M, Chachoua L, Caban A, Toumi M. Managed entry agreements: policy analysis from the European perspective. Value in Health. 2020;23(4):425–33.
National Health Service England. Appraisal and Funding of Caner Drugs from July 2016 (including the new Cancer Drug Fund). A new deal for patient, taxpayers and industry. 08/07/2016 2016. [Online]. https://www.england.nhs.uk/wp-content/uploads/2013/04/cdf-sop.pdf. Accessed 09 Oct 2022.
Wood EM, Hughes DA. The new and non-transparent Cancer Drugs Fund. Pharmacoeconomics. 2020;38(1):1–4.
National Institute of Health and Care Excellence. The Innovative Medicines Fund Principles. Accessed 11 Nov 2022.
Bril A, Canet E. The innovative medicine initiative (IMI). Médecine/Sciences M/S. 2008;24(10):885–90.
Kang J, Cairns J. ‘Don’t think twice, it’s all right’: using additional data to reduce uncertainty regarding oncologic drugs provided through managed access agreements in England. PharmacoEconomics Open. 2022;7:77–91.
Calnan M, Hashem F, Brown P. Still elegantly muddling through? NICE and uncertainty in decision making about the rationing of expensive medicines in England. Int J Health Serv. 2017;47(3):571–94.
National Institute of Health and Care Excellence. NICE health technology evaluations: the manual. National Institute of Health and Care Excellence 31 January 2022 2022. [Online]. Available: https://www.nice.org.uk/process/pmg36/resources/nice-health-technology-evaluations-the-manual-pdf-72286779244741. Accessed 09 Oct 2022.
Kent E, Groves B, Strong T. PMU84 worlds apart or close relations? A comparison of managed access agreements (MAAS) in orphan versus cancer treatments: insights from England. Value in Health. 2020;23:S248.
Adler B, Privolnev Y. PCN276-managed access agreements under the Cancer Drugs Fund (CDF): key metrics and drivers. Value in Health. 2018;21:S61.
National Institute of Health and Care Excellence. National Institute for Health and Care Excellence. 2022. https://www.nice.org.uk/. Accessed 11 Nov 2022.
European Medicines Agency. “European Medicines Agency.” https://www.ema.europa.eu/en/medicines. Accessed 09 Oct 2022.
Medicines and Healthcare Products Regulatory Agency. “MHRA Products." NHRA Products. Accessed 11 Nov 2022.
Attride-Stirling J. Thematic networks: an analytic tool for qualitative research. Qual Res. 2001;1(3):385–405.
Morrell L, Wordsworth S, Barker R. Health technology appraisal and access to cancer drugs in the UK: CASMI full report. Centre for the Advancement of Sustainable Medical Innovation; 2018.
Tai T-A, Latimer NR, Benedict Á, Kiss Z, Nikolaou A. Prevalence of immature survival data for anti-cancer drugs presented to the National Institute for Health and Care Excellence and impact on decision making. Value in Health. 2021;24(4):505–12.
Gov.uk. Innovative Licensing and Access Pathway. https://www.gov.uk/guidance/innovative-licensing-and-access-pathway. Accessed 11 Nov 2022.
Efthymiadou O, Kanavos P. Impact of managed entry agreements on availability of and timely access to medicines: an ex-post evaluation of agreements implemented for oncology therapies in four countries. BMC Health Serv Res. 2022;22(1):1066. https://doi.org/10.1186/s12913-022-08437-w.
Wonder M, Backhouse M, Sullivan S. Australian managed entry scheme: a new managable proccess for the reimbursement of new medicines? Value Health. 2012;15(3):586–90.
Farmer C, O’Toole B, Barnish MS, Trigg LA, Hayward S, Crathorne L, Kasten Z, Melendez-Torres GJ. Early access schemes for innovative health technolgies: the views of international stakeholders. Int J Technol Assess Health Care. 2023;39(1):E45. https://doi.org/10.1017/S0266462323000429.
Acknowledgements
Funding for this project was provided by NHS England.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing Interests
No authors declare any competing interests relevant to the present work.
Funding
This work was supported by NHS England.
Availability of Data and Material.
The study was informed by publicly available data found at https://www.nice.org.uk, and included an analysis of managed access agreements of technologies in the Cancer Drugs Fund, and their corresponding data collection agreements.
Ethics Approval
The project was approved by the University of Exeter College of Medicine and Health Research Ethics committee (ref: 20/11/264).
Author Contributions
CF, BG and JS designed the research. NS identified all relevant source materials. LT, SH, LC and M.B contributed to the data extraction. GMT, CF and LT carried out the data analysis for the uncertainties, and LT carried out the data analysis for the data collection agreements. CF designed the data collection tools, quality assured the data extraction and is the guarantor for this manuscript. LT drafted the manuscript with all authors contributing to the revision. The authors acknowledge the administrative support provided by Mrs Sue Whiffin and Ms Jenny Lowe.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Code Availability:
Not applicable. Data available upon request.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Trigg, L.A., Barnish, M.S., Hayward, S. et al. An Analysis of Uncertainties and Data Collection Agreements in the Cancer Drugs Fund. PharmacoEconomics Open 8, 303–311 (2024). https://doi.org/10.1007/s41669-023-00460-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s41669-023-00460-9