
Abstract
A facile and rapid approach for detecting low concentration of iron ion (Fe3+) with improved sensitivity was developed on the basis of plasmon enhanced fluorescence and subsequently amplified fluorescence quenching. Au1Ag4@SiO2 nanoparticles were synthesized and dispersed into fluorescein isothiocyanate (FITC) solution. The fluorescence of the FITC solution was improved due to plasmon enhanced fluorescence. However, efficient fluorescence quenching of the FITC/Au1Ag4@SiO2 solution was subsequently achieved when Fe3+, with a concentration ranging from 17 nM to 3.4 μM, was added into the FITC/Au1Ag4@SiO2 solution, whereas almost no fluorescence quenching was observed for pure FITC solution under the same condition. FITC/Au1Ag4@SiO2 solution shows a better sensitivity for detecting low concentration of Fe3+ compared to pure FITC solution. The quantized limit of detection toward Fe3+ was improved from 4.6 μM for pure FITC solution to 20 nM for FITC/Au1Ag4@SiO2 solution.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Noble metal nanoparticles (NPs), such as Au and Ag, are known to dramatically change the optical properties of nearby fluorophores due to the localized surface plasmon resonance (LSPR) at the surface of the metal NPs [1]. When fluorescent molecules are localized adjacent to metal surface, their fluorescence emission intensity can be altered enormously, forming the basis of plasmon enhanced fluorescence (PEF) [2]. However, if fluorescent molecules are directly in contact with the metal surface, fluorescence quenching would be suffered due to the non-radiative energy and/or charge transfer from molecules to the metal. A nanometer-thin spacer layer, made of either polyelectrolytes or silica, is usually employed to separate the molecules away from the metal surface to avoid fluorescence quenching [2–4]. The fluorescence of fluorophores can make a continuous transition from fluorescence quenching to fluorescence enhancement with increasing thickness of the silica layer [5–9]. Great attentions have been paid to prepare metal core-silica/fluorophore-shell nanostructure for various applications, such as optical property [7], cellular imaging [10], and photothermal therapy [11]. The excited surface plasmon can deliver significant control over the optical field and enhance the light absorption or fluorescence emission of molecule, which is critical to improving the sensitivity of fluorescence spectroscopy [12].
During the past couple of decades, fluorescence detection offers a promising approach for simple and rapid tracking of heavy metal ions [13, 14]. Contamination of water by metal ions (e.g., mercury, copper, iron) can cause serious environmental and health problems because of acute and/or chronic toxicity to biological organisms [15–17]. Therefore, monitoring of metal ion levels in water is very important for the environment or our health. The conventional detection concentration of metal ion based on fluorophore or conjugated polymer is on the micromolar (μM) level [12, 18]. Therefore, many AuNPs-based colorimetric, fluorescent or refractive index sensor have been developed to detect lower concentration of metal ions through utilizing fluorescent noble metal nanoclusters or metal NPs fluorescently labeled DNA nanohybrids structures [9, 15–17, 19–23]. However, complicated processes together with expensive fluorophores or coupling reagents for labeling the probes and/or target analytes were involved in the preparation of fluorophore conjugated AuNPs probes [24]. Specialized synthetic skills and complicated purification procedures are rather time-consuming and disadvantage for further practical application. Thus, developing a facile, rapid, and label-free strategy for the detection of metal ions is still highly desired.
Fluorescein isothiocyanate (FITC) has many reactive groups, such as isothiocyanate group (–N=C=S), carboxylic group, hydroxyl group, and carbonyl group (Fig. 1). It has been widely used as a fluorescent label to attach to proteins via the amine group or been tailored for various chemical and biological applications due to different attachment groups, high fluorescence intensity, and great photostability [24–26]. FITC molecules can be readily attached to the surface of AuNPs through their isothiocyanate group [25, 27] and the isothiocyanate group of FITC can also be bond with iron ion (Fe3+) to form metal-isothiocyanate complexes [28].

Schematic illustration of the detection of Fe3+ on the basis of fluorescence enhancement and subsequently fluorescence quenching
Herein, we developed a facile and rapid approach to detecting low concentration of Fe3+ with improved sensitivity on the basis of plasmon enhanced fluorescence and subsequently amplified fluorescence quenching. First, the fluorescence of the FITC solution was greatly improved by the addition of Au1Ag4@SiO2 NPs. And then, the fluorescence of solution was dramatically quenched again when Fe3+ was added. The extent of fluorescence quenching for the FITC/Au1Ag4@SiO2 solution toward Fe3+ is apparently larger than that for pure FITC solution, which resulted in the improved sensitivity for detecting low concentration of Fe3+.
2 Experimental Methods
Au1Ag4 alloy NPs were synthesized by reducing HAuCl4 and AgNO3 solution simultaneously with sodium citrate, according to a reported procedure with slight modifications [30]. 6 mL of HAuCl4 aqueous solution (0.01 mol/L) and 24 mL of AgNO3 aqueous solution (0.01 mol/L) were added to 70 mL ultra-filtered water. The mixture was heated to boiling, and then 6 mL of sodium citrate aqueous solution (1 wt%) was injected quickly. The mixture was further boiled for about 60 min and the color of the solution turned from colorless to yellow within 10 min. Subsequently, 6 mL of sodium citrate aqueous solution was injected again. The mixture was kept boiling for 60 min and then cooled to room temperature.
100 mL of as-synthesized Au1Ag4 alloy NPs was centrifuged at 6,500 rpm, and then was redispersed into 3 mL of water. 1 mL of concentrated Au1Ag4 alloy NPs were added into 30 mL of iso-propanol/9 mL of deionized water mixture with vigorous stirring. Different volume of TEOS (10 mM in iso-propanol) was added to the reaction mixture immediately followed by the addition of 0.75 mL of 25 % ammonium hydroxide. The Au1Ag4@SiO2 NPs with different silica thicknesses were made by tuning the volume of TEOS to 1.6, 3.2, 4.8, and 6.0 mL. The reaction solution was kept at 30 °C for 24 h under stirring.
In a typical process, 400 μL Au1Ag4@SiO2 solution was added to 2.5 mL FITC aqueous solution (5 × 10−6 M), then different volumes of Fe3+ solutions were added. In order to eliminate the possibility of contaminations, Fe3+ solutions were prepared by dissolving desired amount of solid FeCl3 (AR) into ultra-filtered water. The mixture was obtained by repeated gentle inversion for 10 s and then was directly measured for fluorescence emission spectra. Ultra-filtered water was used in all experiments.
Transmission electron microscope (TEM) was taken with TecnaiG220, FEI company microscope operated at 200 kV. Ultraviolet–visible spectroscopy (UV-Vis) extinction spectra were measured with Shimadzu UV-3150 spectrophotometer. Fluorescence emission measurements were recorded with a Horiba FluoroMax-4 spectrofluorometer. Fluorescence lifetimes were performed on FL3-TCSPC Fluorescence Spectroscopy (Horiba Jobin–Yvon Inc., France).
3 Results and Discussion
Our strategy for fluorescence enhancement was to disperse as-synthesized Au1Ag4@SiO2 NPs into FITC solution. The fluorescence of the FITC solution was improved with the addition of Au1Ag4@SiO2 NPs due to PEF. However, the field enhancement region of metal NPs are confined in close vicinity to the surface of plasmonic metal NPs and the fluorescence enhancement is very sensitive to the surrounding environment of metal NPs [2], such as the space between fluorescent molecule and metal NPs. Thus, the previous enhanced fluorescence of FITC by the Au1Ag4@SiO2 NPs was dramatically quenched again when Fe3+ was dropped into the FITC/Au1Ag4@SiO2 solution. The fluorescence quenching is possibly attributed to the complexation action between the isothiocyanate group of FITC and Fe3+, which resulted that FITC molecule was away from the field enhancement region of Au1Ag4@SiO2 NPs (Fig. 1).
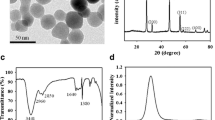
Note that the local electric field enhancement is dependent on the plasmon resonance wavelength of metal NPs. The fluorescence intensity reaches to the maximum when the plasmon wavelength of the metal NPs is between the absorption and emission peak of the fluorescent molecules [29]. However, localized plasmonic resonances have certain peak widths due to damping; the maximal effect is also believed to occur when the emission or excitation peak overlaps closely with the plasmon resonance peak [2]. Only when a plasmon resonance is excited at its peak wavelength can the maximal field enhancement be obtained [2]. We synthesized Au1Ag4 NPs with an extinction peak at 437 nm by the co-reduction of chlorauric acid (HAuCl4) and silver nitrate (AgNO3) solution according to a reported procedure with slight modifications [30]. The average diameter of Au1Ag4 NPs is 35 ± 5 nm. The High-resolution TEM (HRTEM) images of the Au1Ag4 NPs showed the d-spacing for lattice fringes and the corresponding selected area matched well with that of the (111) and (200) planes of face-centered cubic Au and Ag (Fig. 2). High-angle annular dark-field (HAADF) scanning transmission electron microcopy (STEM) image with energy dispersive X-ray spectroscopic (EDX) elemental line profiling and TEM-EDS (TEM-energy dispersive spectrum) of single NPs revealed the alloy structure and composition of NPs (Fig. 2).

a TEM image of Au1Ag4 alloy NPs. b and c High-resolution electron images of Au1Ag4 NPs. d Cross-sectional compositional STEM-EDS line scan profiles recorded from one nanoparticle. Inset is HAADF-STEM image. e TEM-EDS spectrum of a single Au1Ag4 NPs
A SiO2 shell with variable thicknesses was coated on the surface of Au1Ag4 NPs by a sol–gel reaction of TEOS according to the reported process [24]. The fluorescent molecule–metal distance was controlled by varying the thickness of the SiO2 spacer shell. The surface plasmon resonance peak of Au1Ag4@SiO2 NPs red-shifted from 445 to 458 nm as the thickness of the silica shell increased from 6 nm to 33 nm (Fig. 3a–e). The extinction peak of Au1Ag4@SiO2 NPs is close to the absorption peak of the FITC solution (454–474 nm) (Fig. 3f). The silica layer not only provided a distance-dependent PEF but also offered the robustness, chemical inertness, and the versatility needed for the conjugation of fluorescent molecule [4, 7, 31].

TEM images of Au1Ag4@SiO2 with different thicknesses of silica shells: a 6 nm; b 20 nm; c 28 nm; d 33 nm. e The corresponding extinction spectra. f UV–Vis spectrum of the FITC solution
Subsequently, Au1Ag4@SiO2 NPs were directly dispersed into the FITC solution and the influence of Au1Ag4@SiO2 NPs on the fluorescence of the FITC (1 × 10−5 M) were investigated. The emission spectra of the FITC/Au1Ag4@SiO2 solution as well as pure FITC solution are presented in Fig. 4a. The fluorescence of the FITC solution increased greatly as the thickness of the silica shell varied. Maximal fluorescence intensity (about 2.7 times) was observed when the thickness of silica shell was 33 nm. Enhanced fluorescence is also easily discernible to the naked eye under 365 nm irradiation (the photos inset in Fig. 4a). The fluorescence enhancement can also be achieved when decreasing the concentration of FITC aqueous solution. The max fluorescence intensity of the FITC/Au1Ag4@SiO2 solution is 1.9, 2.0, and 3.0 folds of pure FITC solution, corresponding to the FITC solution with a concentration of 5 × 10−6, 1 × 10−7, and 5 × 10−8 M, respectively (Fig. 4b–d).

Fluorescence emission spectra of the FITC solution with the addition of Au1Ag4@SiO2 NPs. The concentration of the FITC solution is: a 1 × 10−5 M; b 5 × 10−6 M; c 1 × 10−7 M and d 5 × 10−8 M, respectively. The volume of the FITC solution is 2.5 mL. The excitation wavelength is 450 nm
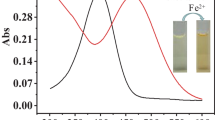
In general, the field-enhanced fluorescence is very sensitive to the surrounding environment of metal NPs. Note that the fluorescence of the solution was varied as the thickness of silica shell changed, we further investigated the metal ion-responsive property of the FITC/Au1Ag4@SiO2 solution. The fluorescence response of the FITC and FITC/Au1Ag4@SiO2 solution toward Fe3+ with a concentration ranging from 17 nM to 63 μM were compared and their fluorescence spectra were recorded (Fig. 5). We found that 34 nM Fe3+ was enough to show efficient fluorescent quenching (14 %) of the FITC/Au1Ag4@SiO2 solution, whereas no fluorescence quenching was observed in pure FITC solution. An apparent fluorescence quenching of pure FITC solution was found until Fe3+ concentration reached 3.4 μM. The fluorescence intensity of the solution kept decreasing as the concentration of Fe3+ increased. About 32 and 68 % of quenching toward 3.4 and 32 μM of Fe3+ were achieved for FITC/Au1Ag4@SiO2 solution, but only about 11 and 43 % of quenching were observed in pure FITC solution under the same condition (Fig. 6). Relative fluorescence intensity ((I0 − I)/I0) of the FITC solution and FITC/Au1Ag4@SiO2 solution toward Fe3+ with a concentration ranging from 17 nM to 63 μM was shown in Fig. 6. The relative fluorescence intensity displayed in Fig. 6, indicated that the extent of fluorescence quenching of the FITC/Au1Ag4@SiO2 solution upon Fe3+ was apparently larger than that in FITC solution and the sensitivity of detecting low concentration of Fe3+ was dramatically improved.

Fluorescence spectra of a FITC and c FITC/Au1Ag4@SiO2 in the presence of Fe3+. b and d The corresponding relationship between intensity ratio (I0/I) and Fe3+ concentration. The excitation wavelength is 450 nm

Relative fluorescence intensity ((I0 − I)/I0) of the FITC solution and FITC/Au1Ag4@SiO2 solution toward Fe3+ with a concentration from 17 nM to 63 μM
The intensity ratio I0/I of pure FITC solution and FITC/Au1Ag4@SiO2 solution displayed a good linear relationship versus Fe3+ concentration ranging from 3.4 to 63 μM, which was easily described by the Stern–Volmer equation (I0/I = 1 + Ksv[Q]), where I0 and I are the fluorescence intensity of the FITC solution in the absence and presence of metal ion, Ksv is the Stern–Volmer fluorescence quenching constant, and Q is the concentration of metal ion. The resulting linear equations were I0/I = 1 + 0.027[Fe3+] and I0/I = 1 + 0.074[Fe3+], corresponding to FITC solution and FITC/Au1Ag4@SiO2 solution, respectively. However, the intensity ratio I0/I for FITC/Au1Ag4@SiO2 solution displayed a different response versus lower concentration of Fe3+ ranging from 17 nM to 3.4 μM. A quantitative analysis of Fe3+ at the lower concentration was achievable (I0/I = 1 + 2.84[Fe3+], R2 = 0.9498). The quantized limit of detection (LOD) toward Fe3+ was calculated to be 20 nM, whereas the LOD value for pure FITC solution toward Fe3+ is only 4.6 μM.
We preliminary investigated the possible reasons for fluorescence enhancement and subsequently fluorescence quenching in this system. Usually, the PEF arises from two contributions. One is the increase in the radiative emission rate, which leads to very short fluorescence lifetime. The other is the increase in the excitation rate due to the local electric field enhancement near the surfaces of metal NPs, which result in surface plasmon enhanced absorption [1, 32, 33]. Figure 7a shows the time-resolved measurements on the emission dynamics of the FITC solution without and in the presence of Au1Ag4@SiO2 NPs. No notable decrease of the fluorescence lifetimes were observed for pure FITC solution and FITC/Au1Ag4@SiO2 solution. The addition of Fe3+ also has no influence on the fluorescent lifetimes. However, the absorption peak of FITC was greatly improved when Au1Ag4@SiO2 NPs were added (Fig. 7b). It is because that plasmonic metal NPs exhibit large absorption and scattering cross sections. So the fluorescence emission from one fluorescence molecule-nanoparticles hybrid nanostructures can be absorbed or scattered by the other hybrid NPs in the solution if the fluorescence emission peak overlaps with the plasmon resonance peak [2]. The fluorescence lifetime data and absorption spectra suggested that the electric field enhanced absorption is the dominant factor in our system. Moreover, the absorption of the FITC/Au1Ag4@SiO2 solution dramatically decreased after the addition of Fe3+ (Fig. 7b), indicating that the aggregation status of FITC molecules changed after the addition of Fe3+. The reason may be attributed to the complexation between isothiocyanate group and Fe3+. Isothiocyanate group of FITC can interact with the Fe3+ to form metal-isothiocyanate complexes [28]. When the Fe3+ was dropped into the FITC solution, bridges would be formed between FITC molecules and possibly induced FITC molecules to aggregate. The aggregation of FITC results in the FITC molecule being dropped away from the silica layer as well as the field enhancement region of NPs and the previously enhanced fluorescence of FITC by Au1Ag4@SiO2 NPs were greatly quenched again due to the promoted distance between FITC molecule and metal NPs.

a The fluorescence intensity decays of FITC and FITC/Au1Ag4@SiO2 solution without or with Fe3+. The instrument response function (IRF) is also included. The concentration of Fe3+ is 3.4 μM. b Absorption spectra of the FITC solution, Au1Ag4 NPs and the FITC solution in the absence or presence of Au1Ag4@SiO2 NPs or Fe3+. The concentration of Fe3+ is 340 nM
4 Conclusions
A facile and rapid approach for detecting low concentration of Fe3+ with improved sensitivity was developed through plasmon enhanced fluorescence and subsequently amplified fluorescent quenching. The fluorescence of the FITC solution was greatly improved by Au1Ag4@SiO2 due to the plasmon enhanced fluorescence. The enhanced fluorescence arises from surface plasmon enhanced absorption. However, efficient fluorescent quenching of the FITC solution was obtained when Fe3+ with a concentration ranging from 17 nM to 3.4 μM was further added into the FITC/Au1Ag4@SiO2 mixture, whereas no fluorescence quenching was observed for pure FITC solution. The amplified fluorescence quenching results in a great increase of sensitivity for detecting low concentration of metal ion. The quantized limit of detection (LOD) was improved from 4.6 μM for pure FITC solution to 20 nM for FITC/Au1Ag4@SiO2 solution. We believe this work would provide a general approach for preparing plasmonic chemsensor with high sensitivity for the detection of low concentration of metal ions.
References
W.H. Ni, Z. Yang, H.J. Chen, L. Li, J.F. Wang, Coupling between molecular and plasmonic resonances in freestanding dye-gold nanorod hybrid nanostructures. J. Am. Chem. Soc. 130(21), 6692–6693 (2008). doi:10.1021/ja8012374
T. Ming, H.J. Chen, R.B. Jiang, Q. Li, J.F. Wang, Plasmon-controlled fluorescence: beyond the intensity enhancement. J. Phys. Chem. Lett. 3(2), 191–202 (2012). doi:10.1021/jz201392k
G. Schneider, G. Decher, Distance-dependent fluorescence quenching on gold nanoparticles ensheathed with layer-by-layer assembled polyelectrolytes. Nano Lett. 6(3), 530–536 (2006). doi:10.1021/nl052441s
P. Reineck, D. Gómez, S.H. Ng, M. Karg, T. Bell, P. Mulvaney, U. Bach, Distance and wavelength dependent quenching of molecular fluorescence by Au@SiO2 core–shell nanoparticles. ACS Nano 7(8), 6636–6648 (2013). doi:10.1021/nn401775e
A.R. Guerrero, R.F. Aroca, Surface-enhanced fluorescence with shell-isolated nanoparticles (SHINEF). Angew. Chem. Int. Ed. 50(3), 665–668 (2011). doi:10.1002/anie.201004806
D.M. Cheng, Q.H. Xu, Separation distance dependent fluorescence enhancement of fluorescein isothiocyanate by silver nanoparticles. Chem. Commun. 3, 248–250 (2007). doi:10.1039/B612401A
K. Aslan, M. Wu, J.R. Lakowicz, C.D. Geddes, Fluorescent core–shell Ag@SiO2 nanocomposites for metal-enhanced fluorescence and single nanoparticle sensing platforms. J. Am. Chem. Soc. 129(6), 1524–1525 (2007). doi:10.1021/ja0680820
J.P. Yang, F. Zhang, Y.R. Chen, S. Qian, P. Hu, W. Li, Y.H. Deng, Y. Fang, L. Han, M. Luqman, D.Y. Zhao, Core–shell Ag@ SiO2@mSiO2 mesoporous nanocarriers for metal-enhanced fluorescence. Chem. Commun. 47(42), 11618–11620 (2011). doi:10.1039/C1CC15304H
C. Wu, J. Zheng, C. Huang, J. Lai, S. Li, C. Chen, Y. Zhao, Hybrid silica-nanocrystal-organic dye superstructures as post-encoding fluorescent probes. Angew. Chem. Int. Ed. 46(28), 5393–5396 (2007). doi:10.1002/anie.200700847
V.W.K. Ng, R. Berti, F. Lesage, A. Kakkar, Gold: a versatile tool for in vivo imaging. J. Mater. Chem. B 1(1), 9–25 (2013). doi:10.1039/C2TB00020B
Z.J. Zhang, J. Wang, C.Y. Chen, Gold nanorods based platforms for light-mediated theranostics. Theranostics 3(3), 223–238 (2012). doi:10.7150/thno.5409
H.M. Su, Y.C. Zhong, T. Ming, J.F. Wang, K.S. Wong, Extraordinary surface plasmon coupled emission using core/shell gold nanorods. J. Phys. Chem. C 116(16), 9259–9264 (2012). doi:10.1021/jp211713y
J. Song, W. Zhang, K.S. Miao, H.L. Zeng, S. Cheng, L.J. Fan, Receptor-free poly (phenylenevinylene) fibrous membranes for cation sensing: high sensitivity and good selectivity achieved by choosing the appropriate polymer matrix. Appl. Mater. Interfaces 5(10), 4011–4016 (2013). doi:10.1021/am4005336
D.Z. Shen, L.S. Wang, Z.X. Pan, S. Cheng, X.L. Zhu, L.J. Fan, Toward a highly sensitive fluorescence sensing system of an amphiphilic molecular rod: facile synthesis and significant solvent-assisted photophysical tunability. Macromolecules 44(4), 1009–1015 (2011). doi:10.1021/ma102311n
G. Sener, L. Uzun, A. Denizli, Lysine promoted colorimetric response of gold nanoparticles: a simple assay for ultra-sensitive mercury (II) detection. Anal. Chem. 86(1), 514–520 (2014). doi:10.1021/ac403447a
T.Y. Zhou, L.P. Lin, M.C. Rong, Y.Q. Jiang, X. Chen, Silver–gold alloy nanoclusters as a fluorescence-enhanced probe for aluminum ion sensing. Anal. Chem. 85(20), 9839–9844 (2013). doi:10.1021/ac4023764
Z.Q. Yuan, N. Cai, Y. Du, Y. He, E.S. Yeung, Sensitive and selective detection of copper ions with highly stable polyethyleneimine-protected silver nanoclusters. Anal. Chem. 86(1), 419–426 (2014). doi:10.1021/ac402158j
X.F. Wu, B.W. Xu, H. Tong, L.X. Wang, Phosphonate-functionalized polyfluorene film sensors for sensitive detection of iron (III) in both organic and aqueous media. Macromolecules 43(21), 8917–8923 (2010). doi:10.1021/ma1019413
X. He, H. Liu, Y. Li, S. Wang, Y. Li, N. Wang, J. Xiao, X. Xu, D. Zhu, Gold nanoparticle-based fluorometric and colorimetric sensing of copper (II) ions. Adv. Mater. 17(23), 2811–2815 (2005). doi:10.1002/adma.200501173
J.P. Xie, Y.G. Zheng, J.Y. Ying, Highly selective and ultrasensitive detection of Hg2+ based on fluorescence quenching of Au nanoclusters by Hg2+-Au+ interactions. Chem. Commun. 46(6), 961–963 (2010). doi:10.1039/B920748A
Y.S. Wu, F.F. Huang, Y.W. Lin, Fluorescent detection of lead in environmental water and urine samples using enzyme mimics of catechin-synthesized Au nanoparticles. Appl. Mater. Interfaces 5(4), 1503–1509 (2013). doi:10.1021/am3030454
J.W. Liu, Y. Lu, A DNAzyme catalytic beacon sensor for paramagnetic Cu2+ ions in aqueous solution with high sensitivity and selectivity. J. Am. Chem. Soc. 129(32), 9838–9839 (2007). doi:10.1021/ja0717358
D.B. Liu, Z. Wang, X.Y. Jiang, Gold nanoparticles for the colorimetric and fluorescent detection of ions and small organic molecules. Nanoscale 3(4), 1421–1433 (2011). doi:10.1039/C0NR00887G
S.C. Wei, P.H. Hsu, Y.F. Lee, Y.W. Lin, C.C. Huang, Selective detection of iodide and cyanide anions using gold-nanoparticle-based fluorescent probes. ACS Appl. Mater. Interfaces 4(5), 2652–2658 (2012). doi:10.1021/am3003044
C.Y. Lin, C.H. Liu, W.L. Tseng, Fluorescein isothiocyanate-capped gold nanoparticles for fluorescent detection of reactive oxygen species based on thiol oxidation and their application for sensing glucose in serum. Anal. Methods 2(11), 1810–1815 (2010). doi:10.1039/C0AY00428F
J.H. Choi, H.S. Kim, J.W. Choi, J.W. Hong, Y.K. Kim, B.K. Oh, A novel Au-nanoparticle biosensor for the rapid and simple detection of PSA using a sequence-specific peptide cleavage reaction. Biosens. Bioelectron. 49, 415–419 (2013). doi:10.1016/j.bios.2013.05.042
B. Hu, Y. Zhao, H.Z. Zhu, S.H. Yu, Selective chromogenic detection of thiol-containing biomolecules using carbonaceous nanospheres loaded with silver nanoparticles as carrier. ACS Nano 5(4), 3166–3171 (2011). doi:10.1021/nn2003053
R. Clark, C.S. Williams, Infra-red spectra (3000–200 cm−1) of metal-isothiocyanate complexes. Spectrochim. Acta 22(6), 1081–1090 (1966). doi:10.1016/0371-1951(66)80198-4
Y.C. Chen, K. Munechika, D.S. Ginger, Dependence of fluorescence intensity on the spectral overlap between fluorophores and plasmon resonant single silver nanoparticles. Nano Lett. 7(3), 690–696 (2007). doi:10.1021/nl062795z
S. Link, Z.L. Wang, M.A. El-Sayed, Alloy formation of gold–silver nanoparticles and the dependence of the plasmon absorption on their composition. J. Phys. Chem. B 103(18), 3529–3533 (1999). doi:10.1021/jp990387w
A.G. Martinez, J.P. Juste, L.M. Liz-Marzan, Recent progress on silica coating of nanoparticles and related nanomaterials. Adv. Mater. 22(11), 1182–1195 (2010). doi:10.1002/adma.200901263
H.J. Chen, T. Ming, L. Zhao, F. Wang, L.D. Sun, J.F. Wang, C.H. Yan, Plasmon-molecule interactions. Nano Today 5(5), 494–505 (2010). doi:10.1016/j.nantod.2010.08.009
O. Stranik, R. Nooney, C. McDonagh, B.D. MacCraith, Optimization of nanoparticle size for plasmonic enhancement of fluorescence. Plasmonics 2(1), 15–22 (2007). doi:10.1007/s11468-006-9020-9
Acknowledgments
This work is supported by the National Natural Science Foundation of China (51003069), Natural Science Foundation of Jiangsu Higher Education Institutions of China (10KJB430014) and A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0/) which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Zhou, L., Zhang, H., Luan, Y. et al. Amplified Detection of Iron Ion Based on Plasmon Enhanced Fluorescence and Subsequently Fluorescence Quenching. Nano-Micro Lett. 6, 327–334 (2014). https://doi.org/10.1007/s40820-014-0005-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40820-014-0005-5