
Abstract
Recent evidence indicates that TBI can increase the risk of developing AUD. TBI and AUD share common symptoms including cognitive dysfunction. Therefore, it is of interest to better understand how reward-mediated behaviors central to alcohol addiction, such as alcohol craving, may interact with the cognitive dysfunction of TBI both at the behavioral and neurobiological level. We also present a preliminary case series as an illustration of how neural activation to alcohol cues may provide insight into the unique brain state of co-occurring mild TBI and AUD. Treatment implications for TBI and AUD and their co-occurrence are also discussed.
Similar content being viewed by others

Introduction
Rates of traumatic brain injury (TBI) and alcohol use disorder (AUD) are high in the USA, particularly among military, veteran, and athlete populations at risk for head injury. Among the civilian population, rates of AUD after TBI are as high as 25 % [1]. Among veterans with TBI, rates of AUD range from 6 to 35 % [2, 3]. Symptoms, cognitive deficits, and neural pathways affected by TBI and AUD often overlap, making these co-occurring conditions challenging to diagnose and treat. This clinical picture is complex because not only is intoxication a risk factor for head injury but also those that experience TBI are at elevated risk for experiencing substance use disorders including AUD [4••, 5••].
In this review, we will describe the clinical picture of co-occurring TBI and AUD, including the overlapping cognitive dysfunction and underlying neurobiological alterations. A neuroimaging case series will be used to illustrate concepts. Finally, we will provide an overview of potential treatments for the co-occurrence of TBI and AUD.
Clinical Picture of Co-occurring TBI and Addiction: From Risk Factor to Vulnerability
Defining TBI and AUD
TBI occurs when a person experiences a physiological disruption of brain function due to an external force. TBI severity is clinically defined by the American Congress of Rehabilitation Medicine [6], the Department of Veterans Affairs/Department of Defense [7], and the Centers for Disease Control and Prevention [8] according to the presence and duration of loss of consciousness (LOC), alteration of consciousness (AOC), and post-traumatic amnesia (PTA). The Glasgow Coma Scale (GCS) score and lesion findings detected by routine structural neuroimaging are also used to characterize TBI severity. Table 1 summarizes how these clinical characteristics define TBI severity into mild, moderate, and severe categories. Mild TBI (mTBI) is defined by a period of LOC lasting less than 30 min or AOC lasting less than 24 h, PTA lasting less than 24 h, a GCS score of 13–15, and an absence of brain lesion findings using standard clinical neuroimaging [6, 9].
AUD ranges in severity from mild to severe and is defined by the American Psychiatric Association in the 5th edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-5) [10] by the presence of at least 2 of the 11 symptoms summarized in Table 2 within a 12-month period. Alcohol abuse and alcohol dependence are severity categories used in the DSM-IV [11] and are no longer used with the DSM-5 [10].
Dual Nature of AUD and TBI Risk
It is well known that alcohol intoxication at the time of injury and/or a history of problem alcohol use is associated with increased risk of incurring a TBI [12, 13]. Alcohol use is also known to negatively impact TBI recovery [14]. Furthermore, recent evidence indicates that TBI is a risk factor for subsequent AUD [4••, 5••, 15]. Miller and colleagues reported that active-duty military personnel with mTBI and no prior AUD or SUD showed increased risk of a new alcohol dependence diagnosis relative to a control group with other injuries [4••]. Risk was significantly different, after controlling for relevant demographic and military characteristics, between two large samples of mTBI (n = 5065) and non-mTBI (n = 44733) active-duty military personnel at all three post-injury time points assessed: 1–30, 31–179, and ≥180 days [4••].
Findings reported by Johnson and colleagues also indicate an increased risk for developing AUD among TBI patients [5••]. In a large sample of active-duty military service members with no prior AUD or SUD, there was an increased incident rate ratio of AUD diagnosis within 1 year of injury among those that incurred a TBI of any severity (n = 53,817) relative to those that did not experience a TBI (n = 151,776) after adjusting for covariates including TBI severity, PTSD, other mental health conditions, and relevant demographic factors [5••]. A subgroup analysis revealed that those experiencing a TBI during deployment had a decreased risk of AUD relative to those without TBI and those experiencing a non-deployment-related TBI had an increased risk of AUD relative to those without TBI [5••]. Johnson and colleagues offer a few explanations for this finding including the following: service members deemed fit to deploy have met rigorous pre-deployment screening and therefore may be more healthy and resilient, fewer service members in the deployment TBI group had a history of positive mental health outcomes (19 %) and prior PTSD (3 %) relative to the non-deployment TBI group (25 % for mental health outcomes and 5 % for PTSD), deployed service members may be subject to stricter alcohol availability policies in theater, and deployed service members also have more contact with the medical system providing greater opportunities for identification, treatment, and intervention [5••]. Therefore, the deployed service member subgroup findings may not be representative of the overall trend. This additional analysis of non-deployment-associated TBI makes these findings more generalizable to civilians. These collective findings demonstrate that there is an increased risk of developing an AUD after TBI even for those without an AUD history. This increased risk is independent of TBI severity, and those incurring a TBI outside of deployment may be at greater risk.
It should be taken into consideration that some recent studies have not found that TBI increases risk for AUD [2, 15]. Most recently, Miles and colleagues (2015) found that mTBI did not predict AUD diagnoses for men or women veterans [15]. This study improved on the Miller and Johnson studies by using a mental health evaluation instead of ICD-9 codes for diagnoses, but the sample size was smaller (N = 1278). Because separate analyses were conducted for men and women in the Miles study [15], it is unknown whether or not mTBI would predict AUD in a pooled sample of men and women. Furthermore, it is not clear whether pre-injury history of AUD, mTBI, PTSD, or other mental health disorders had an impact on findings as these were not accounted for in the study criteria or analyses [15].
While more research is needed on the topic, the collective evidence suggests that those who have experienced a TBI may be more likely to have a premorbid history of AUD or may be more vulnerable for developing subsequent AUD. The heterogeneous nature of TBI may also affect addiction vulnerability. Lesion severity, location, and focality can differ dramatically across individuals and affect morbidity as well as recovery [16, 17]. Lesions located within reward pathways and regions that modulate them may impact the degree to which individuals with TBI may be more or less susceptible to AUD or addiction in general. Previous reports of stroke-induced damage to the insula and basal ganglia, regions important for reward, inducing smoking cessation support this theory [18–20]. Thus, characterization of the heterogeneous neuropathology of TBI through advanced neuroimaging techniques may provide insight into the level of addiction vulnerability or even protection from addiction. Furthermore, understanding potential behavioral and neurobiological explanations for this increased risk or vulnerability can lead to developing effective treatments.
Impact of TBI on Reward-Mediated Behavior
The increased risk for incurring an AUD among the TBI population may be explained by overlapping cognitive deficits that could impact reward-mediated behavior. Below, we examine recent evidence regarding overlapping cognitive deficits in TBI and AUD in an effort to better understand how cognitive dysfunction induced by TBI may have a profound influence on an individual’s reward system and thus how reward-mediated behaviors are expressed for individuals with co-occurring TBI and AUD.
TBI-Induced Cognitive Dysfunction
Cognitive deficits are a hallmark symptom following TBI. Cognitive deficits associated with moderate to severe TBI commonly include memory, executive function, information processing and attention [21]. Cognitive deficits can last for years after significant TBI injury and have a negative impact on life reintegration (i.e., return to work, school, play) [22].
For people with mTBI, cognitive deficits manifest immediately after injury and, for a majority, resolve within 1 to 3 months post-injury [23]. However, for a “miserable minority,” symptoms including cognitive deficits may persist [24]. Evidence suggests non-injury-related comorbidities (e.g., demographic, psychosocial, psychiatric factors) may be strong predictors of the persistence of these prolonged symptoms [25]. A recent systematic review published by the International Collaboration on mTBI Prognosis indicates that cognitive impairments in the following domains are reported to occur within the first 2 weeks of injury: distractibility, attention, memory, verbal learning, information processing speed, and impulsivity [26]. Some of the studies included in this systematic review reported persisting deficits in some of these domains lasting up to 6 months post-injury [26]. However, there was less consistency among the persisting cognitive effects and the authors conclude that large-scale, longitudinal confirmatory studies are necessary to elucidate the most common cognitive domain impairments and recovery course following mTBI [26]. Vanderploeg and colleague (2005) [27] presented data on the long-term neuropsychological consequence of mTBI in a sample of over 4000 veterans with an average post-injury status of 8 years. Theirs was unique in that they had the rare opportunity to match both controls and mTBI groups on preinjury intellectual status. Their findings were consistent with other literature that no adverse long-term neuropsychological effects were present. However, when examining non-traditional ways of characterizing complex attention and working memory, some subtle problems were identified. These findings suggest that while global cognitive functioning recovers in this population, some small alterations may persist.
A recent meta-analysis published by Karr conducted among studies of military blast-induced mTBI corroborates the International Collaboration on mTBI Prognosis systematic review. Karr and colleagues found that the cognitive domains most affected by blast-induced TBI in the post-acute phase were executive function, verbal delayed memory, and processing speed [28]. Notably, the average time post-injury for this mTBI sample was 3.79 years indicating further evidence of persistent cognitive dysfunction [28].
Impulsive behavior is a cognitive domain of interest because of its association with addiction. Studies on impulsivity among mTBI populations are just emerging in the last few years and have included veteran populations with and without co-occurring PTSD. Depue and colleagues report that veterans with co-occurring mTBI and PTSD have reduced anterior amygdala volume relative to controls and that this volume reduction was associated with poor inhibitory control and increased self-reported impulsivity [29]. However, one study demonstrated among veterans with PTSD, mTBI, or co-occurring mTBI and PTSD, those with PTSD self-reported engaging in risky and impulsive behaviors regardless of mTBI diagnosis [30]. Interestingly, no studies to date have examined the cognitive domain of impulsivity using delayed or probability discounting procedures. These procedures classically used in behavioral neuroeconomics may provide additional insight into cognitive dysfunction among the mTBI and co-occurring mTBI and AUD populations. Clearly, further assessments of impulsive behavior among diverse mTBI populations are needed to further understand the potential impact of mTBI on this cognitive domain.
Even in the absence of symptoms including cognitive deficits, experiencing mTBI or even sub-concussive blows alters brain function. This has been determined using multiple neuroimaging modalities including task-based functional magnetic resonance imaging (fMRI), resting state fMRI, and electroencephalography [31••, 32••, 33]. Diffusion tensor imaging (DTI) has also shown some promise in the classification of mTBI and as a potential biomarker of recovery from cytotoxic edema in cerebral white matter [34]. This is relevant because the forces following brain trauma creates a particular mechanical vulnerability for damage to frontotemporolimbic regions, which alters both their function and structure, and thus disrupts cognitive processes mediated by these structures [31••, 35]. Furthermore, alterations in brain function critical to cognitive processes may also be revealed when the body is put under physical stress [36••, 37]. Therefore, it is prudent to use a multi-modal neuroimaging approach to developing biomarkers of the long-term recovery and or sequelae of mTBI [38].
Alcohol Addiction and Reward-Mediated Behavior
In the previous section, AUD was defined with a description of symptoms comprising its diagnosis. Here, we further discuss the processes of alcohol addiction in order to shed light upon how alcohol reward-mediated behaviors may be influenced by TBI-related cognitive dysfunction. There are five classic models of addiction that we will describe here which can be integrated into a framework to understand how reward may be altered with co-occurring TBI and AUD: negative reinforcement [39, 40], positive reinforcement [41], incentive salience [42], impulse control [43, 44], and habit learning [45]. We do not propose one model as a singular explanation of addiction for AUD or co-occurring AUD and TBI. What is presented here and in Fig. 1a is an integration of aspects of these models.

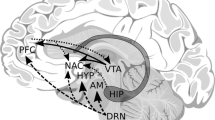
(A) Integrated behavioral/cognitive model of addiction. (B) Blocks of alternating visual alcohol, visual neutral cues, and a craving scale followed by a fixation cross were presented. This blocked cue protocol was modified from that of Vollstädt-Klein and colleagues published in 2010 [46]. (C) The number of activated voxels (AV) in response to the visual alcohol cues minus the visual neutral cues are displayed. The relative t value threshold was set to 50 % of the peak t value for each subject independently
Alcohol addiction is theorized by Koob to be composed of three cyclic phases: preoccupation/anticipation, binge/intoxication, and withdrawal/negative affect [39]. In the negative reinforcement model of addiction, drinking behavior occurs in order to remove internal negative affect. This is delineated from more long-term, external negative consequences that can occur among people with AUD (e.g., legal problems). Internal negative affect is prominent during the withdrawal phase. However, it is important to note that in the course of addiction, relapse to drinking behavior occurs outside of acute withdrawal [47, 48] and treating the withdrawal syndrome alone has not proven sufficient.
Alternatively, the positive reinforcement model is that drinking behavior occurs due to the rewarding or euphoric properties of alcohol [41]. That is, in the negative reinforcement model, the drinking response occurs because removing negative feelings is the reinforcing stimulus. In the positive reinforcement model, the drinking response occurs because of the rewarding/euphoric properties of the alcohol stimulus.
In the incentive salience model [42], alcohol and alcohol-related stimuli sensitize reward pathways in the brain such that “wanting” to drink becomes pathological. Alcohol-related contextual cues become increasingly salient. Therefore, alcohol-related stimuli can elicit the response of drinking behavior. Thus, the stimulus/response relationship of the incentive salience model is in the opposite direction as in negative/positive reinforcement described above (Fig. 1a). That is, with negative and positive reinforcement (as well as impulse dyscontrol described below), the response of drinking leads to a stimulus (e.g., euphoria). However, with incentive salience (as well as habit learning described below), exposure to alcohol-related stimuli leads to a drinking response.
The impulse dyscontrol model includes two systems that are sub-served by defined neural pathways: an impulsive system and a reflective system [43]. The impulsive system transmits immediate signals related to affective or emotional responses. The reflective system transmits long-term signals related to domains such as memory and executive function. Through the reflective system, negative consequences associated with alcohol can be evaluated. Under the impulse dyscontrol model, vulnerability to alcohol addiction can occur when the ability to inhibit the impulsive system is weakened.
The habit learning model builds from the reinforcement and impulse dyscontrol models described above. The habit learning model posits that alcohol use transitions from occurring voluntarily due to largely positive reinforcing experiences to a habitual or compulsive behavior that occurs due to lack of control (e.g., impulse dyscontrol) [45].
Many factors may impact how reward-mediated behaviors are expressed or how drinking behavioral responses occur. Craving, a hallmark symptom of AUD, is one of these factors. Craving is an important symptom to understand because it is a distracting and troubling experience associated with relapse [49] and represents a barrier to recovery. A comprehensive overview of the cognitive perspective on craving is beyond the scope of this review, but has been the subject of previously published reviews [50, 51]. Here, we focus on one perspective craving model, the cognitive processing model developed by Tiffany [52].
The cognitive processing model of craving differentiates itself by viewing alcohol use as a response that can occur in the absence of, or precipitated by, craving [52]. In the cognitive processing model, contextual stimuli can trigger alcohol use through an automatic process in the absence of craving. However, in the nonautomatic process, stimuli trigger behaviors that are met with barriers to drinking, eliciting a craving. Studies in which alcohol related cues slowed reaction time on cognitive tasks support this craving model [50]. People with TBI can experience slowed reaction time among other cognitive dysfunction (described above) as a result of injury. TBI can slow higher cortical function while also disinhibiting motivation and craving associated with addiction. Interestingly, there is some evidence that cognitive rehabilitation treatment administered to people with substance use disorders which is designed to improve cognitive dysfunction can also improve substance use outcomes such as frequency of substance use and addiction severity index measures [53]. It is important to note that craving can occur outside of the circumstances of encountering a barrier to drug or alcohol access. Small priming doses of abused substances produce robust craving responses [54]. Specifically, there is evidence that priming doses of alcohol induce alcohol craving [55, 56]. Exposure to alcohol-related contextual cues [46, 57] as well as stress [58] is also known to elicit craving as well as activation within reward pathways.
What has not been presented before is the integration of these classic addiction models with that of Tiffany’s cognitive processing model in order to provide a framework that may shed light on alcohol addiction for people with co-occurring TBI and AUD. As illustrated in Fig. 1a, (1a) negative and (1b) positive reinforcement as well as (1c) impulse dyscontrol models are illustrated on the left where a behavioral response (i.e., alcohol use) leads to a stimulus (i.e., removal of negative affect/reward). Illustrated on the right are the (3a) incentive salience and (3b) habit learning models in which a generally positive stimulus, such as the rewarding effects of alcohol, leads to a response, such as alcohol use. Thus, the order of response and stimuli is reversed when comparing 1a, 1b, and 1c models relative to 3a and 3b models. This figure illustrates how these models can be used together to provide insight for interpreting reward-mediated behavior in addiction. In situations where automated processes are disrupted by barriers to alcohol access, the link between stimulus and response may be disrupted. This disruption may allow for transitions between phases of these addiction models as well as exacerbation of alcohol craving. For a person with TBI and AUD, negative reinforcement (1a) may be involved due to depression or anxiety symptoms but they may also experience cognitive dysfunction which would create more barriers to alcohol access (2) and perpetuate alcohol craving.
It is important to discuss here that depression and anxiety often co-occur with TBI [59, 60]. Likewise, AUD, depression, and anxiety also co-occur [61, 62]. Alcohol may provide short-term relief from internal negative affective states. However, in the long term, alcohol exacerbates depression and anxiety symptoms which then lead to subsequent relapse [63]. Over time, rewiring of the brain also occurs leading to alterations in how people with addictions perceive reward over time [64]. There is evidence that people with AUD favor smaller immediate rewards over larger delayed rewards [65]. Therefore, the temporal horizon is shortened for people with AUD and it is logical to theorize that a sense of short-term relief of negative affect may be valued more than possible negative long-term consequences that could occur due to drinking.
These circumstantial barriers to alcohol access may stand in for the neurobiological brake that the reflective system provides over the impulsive system drive in the impulse dyscontrol model (1c). However, with a weakened system, the impulsive system may lead to impulsive choices during this period without alcohol access that would increase the likelihood of obtaining alcohol and drinking. These impulsive choices may even result in breaking the law to obtain alcohol. When faced with alcohol-related stimuli during this period where barriers to access are experienced, the drive to obtain alcohol becomes stronger due to the principles of incentive salience (3a). Thus, many principles of these classic addiction models are at play for the person with TBI and AUD. As illustrated in the example above, TBI and associated behavioral sequelae may leave a person more vulnerable to alcohol use or alcohol craving.
The translation of the affective states and craving associated with repeated alcohol use to the engagement in the reward-mediated behavior of relapse for the individual with co-occurring TBI and AUD may be influenced by increased impulsive behavior and decreased executive function. There is evidence that impulsive behavior is associated with the initiation of alcohol use [44], which may help to explain the findings of Miller [4••] and Johnson [5••] that incurring a TBI increases the risk of developing an AUD. Furthermore, self-reported ratings of impulsivity are associated with alcohol use which is described by Lejuez and colleagues to produce a bidirectional relationship between alcohol use and impulsivity [44]. An individual with co-occurring TBI and AUD may be more vulnerable to this bi-directional relationship, thus exacerbating AUD. Individuals struggling with addiction may also experience a hypoactivity of neural systems engaged in top-down inhibitory control involved with decision-making process (i.e., reflective system) or executive function leading to relapse [66]. Thus, executive function, too, may be impaired for the individual with co-occurring TBI and AUD leading to exacerbation of symptoms including alcohol craving and vulnerability to relapse.
Cognitive deficits associated with mTBI overlap with AUD and may explain potential vulnerability of the TBI population to AUD and exacerbation of AUD among individuals with co-occurring TBI and AUD. This exacerbation may include increased craving severity among people with co-occurring TBI and AUD. We have recently reported that veterans with a probable AUD and a combination of mental health disorders with and without mTBI self-reported higher craving levels than veterans with probable AUD alone [67••]. Future studies will determine more specifically the effect of mTBI on alcohol craving levels. Moreover, TBI can have a fundamental impact on experiencing reward. Experiencing a TBI may lead to damages in the brain’s reward system leading to compounding these effects and exacerbating the vicious cycle of addiction. For example, TBI can affect naturally occurring intrinsic rewards such as sex. Sexual dysfunction has been reported as occurring in a substantial number of people with TBI [68–70]. Hyposexuality is the most common form of sexual dysfunction following TBI and can occur at all stages of the sexual response cycle, including arousal, sexual behavior/experience, and orgasm [69]. Hypersexuality or disinhibited sexual behavior can occur following TBI, but it is relatively rare, occurring in less than 10 % of persons with TBI who receive rehabilitation services [71]. Persons with TBI are often socially isolated, and poor social participation is a contributor to sexual impairment in persons with TBI. Consistent with the negative reinforcement model [72], they may drink alcohol to avoid the loneliness and pain associated with social isolation, not being aware that alcohol can have a negative impact on their sexual functioning.
Overlapping Neurobiological Pathways of AUD and mTBI
TBI and associated mental health disorders commonly involve abnormalities within reward pathways that drive both the genesis and continuation of addiction [73–75]. Thus, the increased susceptibility for incurring AUD among people with TBI may be due to an underlying neurobiological phenomenon.
Advanced imaging techniques, such as magnetic resonance imaging (MRI), have been used to study brain volume, neural activation, and neural connectivity of reward pathways. Reward pathways associated with craving and addiction are well-defined for individuals with AUD alone. Comprehensive assessment of reward pathways using structural MRI with advanced morphometric analyses demonstrate that people with AUD have reduced volume relative to control participants in the reward network comprised of the dorsolateral prefrontal cortex, insula, subcallosal, orbitofrontal and cingulate cortices, parahippocampal gyrus, and temporal pole as well as the sub-cortical structures of the nucleus accumbens, amygdala, hippocampus, and ventral diencephalon [76]. Moreover, among the AUD group, decreased reward network volume was significantly associated with decreased working memory scores [76]. Another structural neuroimaging study found that amygdala, hippocampus, and ventral striatum volumes are reduced among individuals with AUD relative to control participants. Increased alcohol craving was significantly associated with reduced amygdala volume among individuals with AUD and this change in volume predicted who would relapse 6 months after an initial 1-week detoxification period [77].
Task-based fMRI protocols in which people are presented with alcohol cues have been repeatedly used to define these pathways [58;78]. For example, brain activation in response to visual alcohol cues within the dorsal striatum is positively associated with craving [46] and within the medial prefrontal cortex (mPFC) is positively associated with relapse [78]. Brain activation in the insula, hippocampus, thalamus, and cingulate cortex is also increased in response to alcohol-related images [78–80]. In addition, mental health disorder symptoms, particularly of depression and anxiety, have been shown to positively correlate with alcohol cue-induced brain activation in the insula, cingulate, striatum, and thalamus among people with AUD [81].
Connectivity between these reward-related brain regions may also be compromised by co-occurring TBI and AUD. Resting state functional connectivity (rsFC) using fMRI is a means by which this can be investigated. Both mTBI [36••, 82–84] and alcohol exposure [85, 86] result in abnormal resting state connectivity in brain networks important for cognition and reward. This is relevant because dysfunction in cognition and reward processing influence craving and relapse. When AUD and TBI co-occur, unique differences in alcohol cue-induced neural activation and connectivity at rest may shed light on their underlying neurobiology and thus dictate development of targeted treatments to reduce alcohol craving and relapse.
Preliminary Case Series
We present here preliminary results from a neuroimaging case series in order to illustrate the effects that co-occurring mTBI and AUD may have on alcohol cue-induced neural activity. Neural activation was compared among three adult males including a civilian with probable AUD only (case 1), a veteran with mTBI only (case 2), and a veteran with co-occurring probable AUD and mTBI (case 3). mTBI was defined using a structured clinical interview based on the American Congress of Rehabilitation Medicine definition of mTBI [6] along with neuropsychological test performance measures [87]. Probable AUD was defined as a positive screen on the alcohol use disorder identification test consumption questions (Audit-C; score of ≥4 for men indicates probable AUD) [88]. fMRI data were acquired in the presence of alcohol cues (Fig. 1b) via a neuroimaging protocol adapted by Volstädt-Klein [46]. The veteran with co-occurring mTBI and AUD (case 3) had the greatest brain activation in response to alcohol cues (Fig. 1c).
Region-of-interest analyses demonstrated that the location of alcohol cue-induced activation within the reward pathway was different for each of the three subjects. Activation for the veteran with mTBI only (case 2) had little to no activation with the alcohol reward pathway as expected. Activation for case 1, with AUD only, was located within multiple regions of the reward pathway (e.g., mPFC, putamen, caudate, and insula), but the majority was located within the mPFC and there was no activation within the dorsal cingulate cortex (dCC). Activation for case 3, the veteran with co-occurring mTBI and AUD, is unique in that the activation to alcohol cues was only located within the dCC of the reward pathway, which plays an important role in executive functioning and error processing [89]. The dCC is also part of a network that processes value or importance of reward (i.e., the salience network) [90]. Activation within the entire cingulate cortex, in fact, has been associated with alcohol craving and relapse [91•]. These preliminary findings are the first to examine neural activation in response to alcohol cues among an individual with co-occurring mTBI and AUD. Collectively, these findings suggests that the dCC may be more susceptible to responding to alcohol cues for people with co-occurring conditions whereas the mPFC may be more susceptible to responding to alcohol cues for people with AUD alone.
Informing Treatments to Address Co-occurring TBI and Addiction
There are limited treatment options specifically tailored to individuals with co-occurring TBI and AUD. However, our enhanced understanding of behavioral and neurobiological characteristics of TBI and AUD can be used to guide clinicians on the use of existing treatments for individuals with co-occurring TBI and AUD. Below, we discuss potential behavioral, pharmacological, and neuromodulatory treatments for these conditions.
Behavioral
In treating people with co-occurring TBI and AUD, it is important to provide individualized treatment. Behavioral symptoms of alcohol withdrawal, particularly in early abstinence, include impulsivity, anxiety, irritability, dysphoria, insomnia, and impaired concentration [92]. These symptoms overlap with the emotional and cognitive dysfunction resulting from TBI as described above [93]. Implementing multiple treatments to treat an individual’s specific symptoms may prove inefficient. Thus, utilizing treatments that target multiple symptoms is important for people with co-occurring conditions. One promising treatment is exercise. Exercise is an established intervention for depression, anxiety, and fatigue and improves global cognitive functioning [94]. Engaging in exercise on a routine basis facilitates neurogenesis and neuroplasticity [94–96]. Neuroplasticity, in particular, decreases neuroinflamation and neurodegeneration that may be contributing to the neurobiological deficits of both AUD and TBI. This patient population has a broad array of symptom complexity and often co-occurring mental health conditions. Therefore, pairing an exercise regimen with effective treatment targeted towards symptom presentation may promote an optimal outcome.
The research on effectiveness of treatment for alcohol abuse following TBI is in its infancy. Due to cognitive dysfunction, people with TBI may have difficulty benefitting from traditional substance abuse treatments, such as 12-step programs. Adaptations to existing treatments may be needed, including repetition, concrete examples, use of visual aids, peer-modeling, and slower pace of instruction. Results of recent studies have indicated that brief interventions combining motivational interviewing with education about the negative impact of substance use on TBI recovery show promise for altering expectations about alcohol use [97••] and for decreasing alcohol use [98] in people with TBI; however, studies are methodologically limited and further research on the effectiveness of these interventions for changing alcohol use is needed.
Pharmacological
There are three FDA-approved medications for alcohol dependence, all of which have good evidence and can be safely used in patients with TBI: disulfiram, acamprosate, and naltrexone. The aversion therapy of disulfiram works for patients committed to complete sobriety. One small study of adolescents shows higher abstinence rates and duration [99]. A disadvantage is that disulfiram does not prevent impulsivity and craving resulting in treatment failures. Acamprosate has few side effects and increases abstinence duration [99]. Acamprosate requires good compliance, is dosed three times daily, and should be avoided in the moderately renal impaired. Naltrexone reduces the number of heavy drinking days [99]. Naltrexone can be dosed once daily orally or once monthly by injection, which is beneficial for patients with problems of compliance, memory, and impulsivity. It has high tolerability but should be avoided in patients with liver dysfunction and those who are also prescribed opiates.
There are a number of potential pharmacological treatments that provide promise for treating AUD. These are briefly reviewed here with consideration for patients with co-occurring AUD and TBI.
High-dose gabapentin, 1800 mg/day, has shown better efficacy for AUD than gabapentin 900 mg/day. Increase in abstinence rates, reduced heavy drinking days, and fewer relapse-related symptoms of insomnia, dysphoria, and cue-induced alcohol craving were reported [100]. Patients taking gabapentin also showed relief of sleep disturbance, irritability, concentration problems, anxiety, and dysphoria during the protracted withdrawal phase [92].
Trials of the combination of naltrexone and gabapentin show superior efficacy to either agent alone for reducing cravings and number of drinking days, increasing time to relapse, and decreasing sleep disruption [92]. Both have good tolerability and safety profiles.
There is good evidence that mirtazapine can be used for treating both behavioral symptoms and cravings. Several studies of mirtazapine indicate it strengthens impulse control, reduces alcohol cravings and consumption, and has positive effects on sleep and mood [101]. Positive results were observed in dose ranges of 30–60 mg in heavy drinking males [102•]. In a 2-year follow-up study, patients with AUD and depression reported longer abstinence and greater remission in mood symptoms with mirtazapine 30 mg relative to placebo [101].
Topiramate has evidence in multiple trials for abstinence and irritability, hostility, and impulsivity. Doses of 100–300 mg/day improve abstinence up to 6 weeks after detoxification. Patients experienced reduction in anxiety, hostility, and obsessive-compulsivity [103•]. In a study comparing topiramate 300 mg, naltrexone 50 mg, and placebo, patients had longer abstinence and fewer heavy drinking days with topiramate over placebo and no significant difference with naltrexone. Many clinicians may not prefer topiramate for their patients with TBI because (1) it requires long titration to therapeutic dose and (2) has more reported side effects over other agents, including impaired memory and concentration, psychomotor slowing, and dizziness [99], all which can complicate the symptoms of AUD and TBI.
Prazosin has evidence for treatment of hypervigilance and sleep disruption in patients suffering from PTSD. Evidence shows that high doses (16 mg) limits stress-induced cravings and offers control of impulsivity of alcohol consumption [104•] which benefits in the treatment of TBI, as well.
In medications studied thus far, not one single agent has proved effective for all patients with the co-occurring diagnosis of AUD and TBI. Clinical judgment, medical comorbidities, and future trials will guide which agents are used to treat individuals with these chronic co-occurring disorders.
Neuromodulation
Given that TBI and AUD affect overlapping neural pathways, neuromodulatory treatment through non-invasive neural stimulation, such as transcranial magnetic stimulation, may be an ideal treatment for people with co-occurring TBI and AUD. We recently published a review on this topic [105•]. It should also be mentioned that transcranial direct current stimulation is another non-invasive neuromodulatory treatment option that has shown promise for the treatment of alcohol craving and cognitive dysfunction [106–108].
Conclusions
TBI may be a risk factor for developing substance use disorders including AUD. This vulnerability may be explained by overlapping cognitive dysfunction including increased impulsive behaviors and impaired executive function. These cognitive domains engage the same neural pathways integral to reward-mediated behaviors including alcohol craving. This vulnerability may also be altered by the heterogeneous nature of TBI such that lesion properties may dictate whether an individual is more or less susceptible to addiction or if addiction susceptibility is affected at all. Thus, individuals with co-occurring TBI and AUD may have a unique brain state as illustrated in the case series described above (Fig. 1c). The co-occurrence of these conditions, the variance in severity of AUD, as well as the heterogeneity of TBI underscore the need to obtain a comprehensive assessment of this brain state through advanced neuroimaging. A better understanding of this cognitive dysfunction and how reward-mediated behaviors may be exacerbated for the individual with co-occurring TBI and AUD will aid the development of treatments tailored to these individuals.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Ponsford J, Whelan-Goodinson R, Bahar-Fuchs A. Alcohol and drug use following traumatic brain injury: a prospective study. Brain Inj. 2007;21:1385–92.
Heltemes KJ, Dougherty AL, MacGregor AJ, Galarneau MR. Alcohol abuse disorders among U.S. service members with mild traumatic brain injury. Mil Med. 2011;176:147–50.
Cernich AN, Chandler L, Scherdell T, Kurtz S. Assessment of co-occurring disorders in veterans diagnosed with traumatic brain injury. J Head Trauma Rehabil. 2012;27:253–60.
Miller SC, Baktash SH, Webb TS, Whitehead CR, Maynard C, Wells TS, et al. Risk for addiction-related disorders following mild traumatic brain injury in a large cohort of active-duty U.S. Airmen. Am J Psychiatry. 2013;170:383–90. This epidemiological study is one of the first to demonstrate that those experiencing mTBI have an elevated risk of developing a substance use disorder including AUD.
Johnson LA, Eick-Cost A, Jeffries V, Russell K, Otto JL. Risk of alcohol use disorder or other drug use disorder among U.S. Service members following traumatic brain injury, 2008–2011. Mil Med. 2015;180:208–15. This epidemiological study validates the findings of Miller.
ACRM. Definition of mild traumatic brain injury. J Head Trauma Rehabil. 1993;8:86–7.
The Management of Concussion/mTBI Working Group: VA/DoD Clinical Practice Guideline for Management of Concussion / Mild Traumatic Brain Injury; The office of Quality and Performance and Quality Management Directorate, United States Army MEDCOM, 2009.
Centers for Disease Control and Prevention: Report to Congress on Traumatic Brain Injury in the United States: Epidemiology and Rehabilitation; Atlanta, GA, National Center for Injury Prevention and Control: Division of Unintentional Injury Prevention, 2014.
VA/DoD Clinical Practice Guideline for Management of Concussion/Mild Traumatic Brain Injury: J Rehabil Res Dev 2009;46:CP1-68.
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders DSM-V. 5th ed. Washington, D.C: American Psychiatric Association; 2013.
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders DSM-IV-TR. ed Fourth Edition, Text Revision. Washington, D.C: American Psychiatric Association; 2004.
Roozenbeek B, Maas AI, Menon DK. Changing patterns in the epidemiology of traumatic brain injury. Nat Rev Neurol. 2013;9:231–6.
Parry-Jones BL, Vaughan FL, Miles CW. Traumatic brain injury and substance misuse: a systematic review of prevalence and outcomes research (1994-2004). Neuropsychol Rehabil. 2006;16:537–60.
Corrigan JD. Substance abuse as a mediating factor in outcome from traumatic brain injury. Arch Phys Med Rehabil. 1995;76:302–9.
Miles SR, Graham DP, Teng EJ. Examining the influence of mild traumatic brain injury and posttraumatic stress disorder on alcohol use disorder in OEF/OIF veterans. Mil Med. 2015;180:45–52.
Pape T. Acute neurorehabilitation for disorders of consciousness. In: Selzer ME, Clarke S, Cohen LG, Kwakkel G, Miller RH, editors. The textbook of neural repair and rehabilitation. Cambridge, U.K.: Cambridge University Press; 2014. p. 385–404.
Povlishock JT, Katz DI. Update of neuropathology and neurological recovery after traumatic brain injury. J Head Trauma Rehabil. 2005;20:76–94.
Gaznick N, Tranel D, McNutt A, Bechara A. Basal ganglia plus insula damage yields stronger disruption of smoking addiction than basal ganglia damage alone. Nicotine Tob Res. 2014;16:445–53.
Jarraya B, Brugieres P, Tani N, Hodel J, Grandjacques B, Fenelon G, et al. Disruption of cigarette smoking addiction after posterior cingulate damage. J Neurosurg. 2010;113:1219–21.
Suner-Soler R, Grau A, Gras ME, Font-Mayolas S, Silva Y, Davalos A, et al. Smoking cessation 1 year poststroke and damage to the insular cortex. Stroke. 2012;43:131–6.
Dikmen SS, Corrigan JD, Levin HS, Machamer J, Stiers W, Weisskopf MG. Cognitive outcome following traumatic brain injury. J Head Trauma Rehabil. 2009;24:430–8.
Moriarty H, Winter L, Robinson K, True G, Piersol C, Vause-Earland T, et al. Exploration of individual and family factors related to community reintegration in veterans with traumatic brain injury. J Am Psychiatr Nurses Assoc. 2015;21:195–211.
Iverson GL. Outcome from mild traumatic brain injury. Curr Opin Psychiatry. 2005;18:301–17.
Vanderploeg RD, Belanger HG, Curtiss G. Mild traumatic brain injury and posttraumatic stress disorder and their associations with health symptoms. Arch Phys Med Rehabil. 2009;90:1084–93.
McCrea M, Iverson GL, McAllister TW, Hammeke TA, Powell MR, Barr WB, et al. An integrated review of recovery after mild traumatic brain injury (MTBI): implications for clinical management. Clin Neuropsychol. 2009;23:1368–90.
Carroll LJ, Cassidy JD, Cancelliere C, Cote P, Hincapie CA, Kristman VL, et al. Systematic review of the prognosis after mild traumatic brain injury in adults: cognitive, psychiatric, and mortality outcomes: results of the International Collaboration on Mild Traumatic Brain Injury Prognosis. Arch Phys Med Rehabil. 2014;95:S152–73.
Vanderploeg RD, Curtiss G, Belanger HG. Long-term neuropsychological outcomes following mild traumatic brain injury. J Int Neuropsychol Soc. 2005;11:228–36.
Karr JE, Areshenkoff CN, Duggan EC, Garcia-Barrera MA. Blast-related mild traumatic brain injury: a Bayesian random-effects meta-analysis on the cognitive outcomes of concussion among military personnel. Neuropsychol Rev. 2014;24:428–44.
Depue BE, Olson-Madden JH, Smolker HR, Rajamani M, Brenner LA, Banich MT. Reduced amygdala volume is associated with deficits in inhibitory control: a voxel- and surface-based morphometric analysis of comorbid PTSD/mild TBI. Biomed Res Int. 2014;2014:691505.
James LM, Strom TQ, Leskela J. Risk-taking behaviors and impulsivity among veterans with and without PTSD and mild TBI. Mil Med. 2014;179:357–63.
Talavage TM, Nauman EA, Breedlove EL, Yoruk U, Dye AE, Morigaki KE, et al. Functionally-detected cognitive impairment in high school football players without clinically-diagnosed concussion. J Neurotrauma. 2014;31:327–38. This study demonstrates that even sub-concussive hits as experienced in high school football can have deleterious effects on cognition and brain function.
Johnson B, Neuberger T, Gay M, Hallett M, Slobounov S. Effects of subconcussive head trauma on the default mode network of the brain. J Neurotrauma. 2014;31:1907–13. This study demonstrates that even sub-concussive hits as experienced in collegiate rugby players affects brain network activity.
Teel EF, Ray WJ, Geronimo AM, Slobounov SM. Residual alterations of brain electrical activity in clinically asymptomatic concussed individuals: an EEG study. Clin Neurophysiol. 2014;125:703–7.
Mayer AR, Ling J, Mannell MV, Gasparovic C, Phillips JP, Doezema D, et al. A prospective diffusion tensor imaging study in mild traumatic brain injury. Neurology. 2010;74:643–50.
Bigler ED. Traumatic brain injury, neuroimaging, and neurodegeneration. Front Hum Neurosci. 2013;7:395.
Slobounov SM, Gay M, Zhang K, Johnson B, Pennell D, Sebastianelli W, et al. Alteration of brain functional network at rest and in response to YMCA physical stress test in concussed athletes: RsFMRI study. Neuroimage. 2011;55:1716–27. This study demonstrates that even after concussive symptoms have resolved, functional brain networks are altered when the body is subjected to stress.
Zhang K, Johnson B, Gay M, Horovitz SG, Hallett M, Sebastianelli W, et al. Default mode network in concussed individuals in response to the YMCA physical stress test. J Neurotrauma. 2012;29:756–65.
Bailes J, Bravo S, Breiter H, Kaufman D, Lu Z, Molfese D, et al. A call to arms: the need to create an inter-institutional concussion neuroimaging consortium to discover clinically relevant diagnostic biomarkers and develop evidence-based interventions to facilitate recovery. Dev Neuropsychol. 2015;40:59–62.
Koob GF. Theoretical frameworks and mechanistic aspects of alcohol addiction: alcohol addiction as a reward deficit disorder. Curr Top Behav Neurosci. 2013;13:3–30.
Koob GF, Le MM. Addiction and the brain antireward system. Annu Rev Psychol. 2008;59:29–53.
Wise RA. Neurobiology of addiction. Curr Opin Neurobiol. 1996;6:243–51.
Robinson TE, Berridge KC. Addiction. Annu Rev Psychol. 2003;54:25–53.
Bechara A. Decision making, impulse control and loss of willpower to resist drugs: a neurocognitive perspective. Nat Neurosci. 2005;8:1458–63.
Lejuez CW, Magidson JF, Mitchell SH, Sinha R, Stevens MC, de Wit H. Behavioral and biological indicators of impulsivity in the development of alcohol use, problems, and disorders. Alcohol Clin Exp Res. 2010;34:1334–45.
Everitt BJ, Belin D, Economidou D, Pelloux Y, Dalley JW, Robbins TW. Review: Neural mechanisms underlying the vulnerability to develop compulsive drug-seeking habits and addiction. Philos Trans R Soc Lond B Biol Sci. 2008;363:3125–35.
Vollstadt-Klein S, Wichert S, Rabinstein J, Buhler M, Klein O, Ende G, et al. Initial, habitual and compulsive alcohol use is characterized by a shift of cue processing from ventral to dorsal striatum. Addiction. 2010;105:1741–9.
Moos RH, Moos BS. Rates and predictors of relapse after natural and treated remission from alcohol use disorders. Addiction. 2006;101:212–22.
Angres D, Bologeorges S, Chou J. A two year longitudinal outcome study of addicted health care professionals: an investigation of the role of personality variables. Subst Abus. 2013;7:49–60.
Chakravorty S, Kuna ST, Zaharakis N, O’Brien CP, Kampman KM, Oslin D. Covariates of craving in actively drinking alcoholics. Am J Addict. 2010;19:450–7.
Tiffany ST. Cognitive concepts of craving. Alcohol Res Health. 1999;23:215–24.
Tiffany ST, Conklin CA. A cognitive processing model of alcohol craving and compulsive alcohol use. Addiction. 2000;95 Suppl 2:S145–53.
Tiffany ST. A cognitive model of drug urges and drug-use behavior: role of automatic and nonautomatic processes. Psychol Rev. 1990;97:147–68.
Fals-Stewart W, Lam WK. Computer-assisted cognitive rehabilitation for the treatment of patients with substance use disorders: a randomized clinical trial. Exp Clin Psychopharmacol. 2010;18:87–98.
Elman I, Karlsgodt KH, Gastfriend DR, Chabris CF, Breiter HC. Cocaine-primed craving and its relationship to depressive symptomatology in individuals with cocaine dependence. J Psychopharmacol. 2002;16:163–7.
Courtney KE, Ghahremani DG, Ray LA. The effect of alcohol priming on neural markers of alcohol cue-reactivity. Am J Drug Alcohol Abuse. 2015;41:300–8.
Schoenmakers TM, Wiers RW. Craving and attentional bias respond differently to alcohol priming: a field study in the pub. Eur Addict Res. 2010;16:9–16.
Vollstadt-Klein S, Loeber S, Kirsch M, Bach P, Richter A, Buhler M, et al. Effects of cue-exposure treatment on neural cue reactivity in alcohol dependence: a randomized trial. Biol Psychiatry. 2011;69:1060–6.
Seo D, Sinha R. The neurobiology of alcohol craving and relapse. Handb Clin Neurol. 2014;125:355–68.
Koponen S, Taiminen T, Hiekkanen H, Tenovuo O. Axis I and II psychiatric disorders in patients with traumatic brain injury: a 12-month follow-up study. Brain Inj. 2011;25:1029–34.
Zgaljardic DJ, Seale GS, Schaefer LA, Temple RO, Foreman J, Elliott TR. Psychiatric Disease and Post-Acute Traumatic Brain Injury. J Neurotrauma 2015.
Anthenelli RM, Schuckit MA. Affective and anxiety disorders and alcohol and drug dependence: diagnosis and treatment. J Addict Dis. 1993;12:73–87.
Grant BF, Stinson FS, Dawson DA, Chou SP, Dufour MC, Compton W, et al. Prevalence and co-occurrence of substance use disorders and independent mood and anxiety disorders: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Arch Gen Psychiatry. 2004;61:807–16.
Schellekens AF, de Jong CA, Buitelaar JK, Verkes RJ. Co-morbid anxiety disorders predict early relapse after inpatient alcohol treatment. Eur Psychiatry. 2015;30:128–36.
Bickel WK, Miller ML, Yi R, Kowal BP, Lindquist DM, Pitcock JA. Behavioral and neuroeconomics of drug addiction: competing neural systems and temporal discounting processes. Drug Alcohol Depend. 2007;90 Suppl 1:S85–91.
MacKillop J, Miranda Jr R, Monti PM, Ray LA, Murphy JG, Rohsenow DJ, et al. Alcohol demand, delayed reward discounting, and craving in relation to drinking and alcohol use disorders. J Abnorm Psychol. 2010;119:106–14.
Goldstein RZ, Volkow ND. Drug addiction and its underlying neurobiological basis: neuroimaging evidence for the involvement of the frontal cortex. Am J Psychiatry. 2002;159:1642–52.
Herrold AA, Jordan N, High WM, Babcock-Parziale J, Chambers RA, Smith B, et al. Alcohol use and craving among veterans with mental health disorders and mild traumatic brain injury. J Rehabil Res Dev. 2014;51:1397–410. This study represents the necessary first step in examining alcohol craving among people with co-occurring mTBI, AUD and mental health disorders. Findings demonstrated that veterans with probable AUD and mental health disorders with and without mTBI demonstrated elevated alcohol craving levels relative to veterans with probable AUD alone.
Ponsford J. Sexual changes associated with traumatic brain injury. Neuropsychol Rehabil. 2003;13:275–89.
Sander AM, Maestas KL, Pappadis MR, Sherer M, Hammond FM, Hanks R. Sexual functioning 1 year after traumatic brain injury: findings from a prospective traumatic brain injury model systems collaborative study. Arch Phys Med Rehabil. 2012;93:1331–7.
Hanks RA, Sander AM, Millis SR, Hammond FM, Maestas KL. Changes in sexual functioning from 6 to 12 months following traumatic brain injury: a prospective TBI model system multicenter study. J Head Trauma Rehabil. 2013;28:179–85.
Simpson GK, Sabaz M, Daher M. Prevalence, clinical features, and correlates of inappropriate sexual behavior after traumatic brain injury: a multicenter study. J Head Trauma Rehabil. 2013;28:202–10.
Sander AM, Maestas KL, Nick TG, Pappadis MR, Hammond FM, Hanks RA, et al. Predictors of sexual functioning and satisfaction 1 year following traumatic brain injury: a TBI model systems multicenter study. J Head Trauma Rehabil. 2013;28:186–94.
Koob GF. Neurocircuitry of alcohol addiction: synthesis from animal models. Handb Clin Neurol. 2014;125:33–54.
Breiter HC, Rosen BR. Functional magnetic resonance imaging of brain reward circuitry in the human. Ann N Y Acad Sci. 1999;877:523–47.
Chambers RA, Krystal JH, Self DW. A neurobiological basis for substance abuse comorbidity in schizophrenia. Biol Psychiatry. 2001;50:71–83.
Makris N, Oscar-Berman M, Jaffin SK, Hodge SM, Kennedy DN, Caviness VS, et al. Decreased volume of the brain reward system in alcoholism. Biol Psychiatry. 2008;64:192–202.
Wrase J, Makris N, Braus DF, Mann K, Smolka MN, Kennedy DN, et al. Amygdala volume associated with alcohol abuse relapse and craving. Am J Psychiatry. 2008;165:1179–84.
Grusser SM, Wrase J, Klein S, Hermann D, Smolka MN, Ruf M, et al. Cue-induced activation of the striatum and medial prefrontal cortex is associated with subsequent relapse in abstinent alcoholics. Psychopharmacology (Berl). 2004;175:296–302.
George MS, Anton RF, Bloomer C, Teneback C, Drobes DJ, Lorberbaum JP, et al. Activation of prefrontal cortex and anterior thalamus in alcoholic subjects on exposure to alcohol-specific cues. Arch Gen Psychiatry. 2001;58:345–52.
Hermann D, Smolka MN, Wrase J, Klein S, Nikitopoulos J, Georgi A, et al. Blockade of cue-induced brain activation of abstinent alcoholics by a single administration of amisulpride as measured with fMRI. Alcohol Clin Exp Res. 2006;30:1349–54.
Feldstein Ewing SW, Filbey FM, Chandler LD, Hutchison KE. Exploring the relationship between depressive and anxiety symptoms and neuronal response to alcohol cues. Alcohol Clin Exp Res. 2010;34:396–403.
McDonald BC, Saykin AJ, McAllister TW. Functional MRI of mild traumatic brain injury (mTBI): progress and perspectives from the first decade of studies. Brain Imaging Behav. 2012;6:193–207.
Stevens MC, Lovejoy D, Kim J, Oakes H, Kureshi I, Witt ST. Multiple resting state network functional connectivity abnormalities in mild traumatic brain injury. Brain Imaging Behav. 2012;6:293–318.
Johnson B, Zhang K, Gay M, Horovitz S, Hallett M, Sebastianelli W, et al. Alteration of brain default network in subacute phase of injury in concussed individuals: resting-state fMRI study. Neuroimage. 2012;59:511–8.
Santhanam P, Coles CD, Li Z, Li L, Lynch ME, Hu X. Default mode network dysfunction in adults with prenatal alcohol exposure. Psychiatry Res. 2011;194:354–62.
Roussotte FF, Rudie JD, Smith L, O’Connor MJ, Bookheimer SY, Narr KL, et al. Frontostriatal connectivity in children during working memory and the effects of prenatal methamphetamine, alcohol, and polydrug exposure. Dev Neurosci. 2012;34:43–57.
Pape T, Herrold AA, Smith B, Babcock-Parziale J, Harp J, Shandera-Ochsner A et al. Algorithm for symptom attribution and classification following possible mild traumatic brain injury. J Head Trauma Rehabil 2015;under review.
Bush K, Kivlahan DR, McDonell MB, Fihn SD, Bradley KA. The AUDIT alcohol consumption questions (AUDIT-C): an effective brief screening test for problem drinking. Ambulatory Care Quality Improvement Project (ACQUIP). Alcohol Use Disorders Identification Test. Arch Intern Med. 1998;158:1789–95.
Haber SN, Knutson B. The reward circuit: linking primate anatomy and human imaging. Neuropsychopharmacology. 2010;35:4–26.
Seeley WW, Menon V, Schatzberg AF, Keller J, Glover GH, Kenna H, et al. Dissociable intrinsic connectivity networks for salience processing and executive control. J Neurosci. 2007;27:2349–56.
Schacht JP, Anton RF, Myrick H. Functional neuroimaging studies of alcohol cue reactivity: a quantitative meta-analysis and systematic review. Addict Biol. 2013;18:121–33. This is a very comprehensive and informative review that is very valuable in brining together the collective functional MRI literature using alcohol cues.
Anton RF, Myrick H, Wright TM, Latham PK, Baros AM, Waid LR, et al. Gabapentin combined with naltrexone for the treatment of alcohol dependence. Am J Psychiatry. 2011;168:709–17.
Bales JW, Wagner AK, Kline AE, Dixon CE. Persistent cognitive dysfunction after traumatic brain injury: a dopamine hypothesis. Neurosci Biobehav Rev. 2009;33:981–1003.
Silverberg ND, Iverson GL. Is rest after concussion “the best medicine?”: recommendations for activity resumption following concussion in athletes, civilians, and military service members. J Head Trauma Rehabil. 2013;28:250–9.
Hotting K, Roder B. Beneficial effects of physical exercise on neuroplasticity and cognition. Neurosci Biobehav Rev. 2013;37:2243–57.
Svensson M, Lexell J, Deierborg T. Effects of physical exercise on neuroinflammation, neuroplasticity, neurodegeneration, and behavior: what we can learn from animal models in clinical settings. Neurorehabil Neural Repair. 2015;29:577–89.
Sander AM, Bogner J, Nick TG, Clark AN, Corrigan JD, Rozzell M. A randomized controlled trial of brief intervention for problem alcohol use in persons with traumatic brain injury. J Head Trauma Rehabil. 2012;27:319–30. This is one of the first studies to examine a behavioral treatment intervention specifically for people with co-occurring AUD and TBI. This study demonstrates that a brief intervention of motivational interviewing and education holds promise to change expectations about alcohol use among people with co-occurring AUD and TBI.
Tweedly L, Ponsford J, Lee N. Investigation of the effectiveness of brief interventions to reduce alcohol consumption following traumatic brain injury. J Head Trauma Rehabil. 2012;27:331–41.
Muller CA, Geisel O, Banas R, Heinz A. Current pharmacological treatment approaches for alcohol dependence. Expert Opin Pharmacother. 2014;15:471–81.
Mason BJ, Quello S, Goodell V, Shadan F, Kyle M, Begovic A. Gabapentin treatment for alcohol dependence: a randomized clinical trial. JAMA Intern Med. 2014;174:70–7.
Cornelius JR, Douaihy AB, Clark DB, Daley DC, Chung TA, Wesesky MA et al. Mirtazapine in Comorbid Major Depression and Alcohol Use Disorder: A Long-Term Follow-Up Study. J Addict Behav Ther Rehabil 2013;3.
de Bejczy A, Soderpalm B. The effects of mirtazapine versus placebo on alcohol consumption in male high consumers of alcohol: a randomized, controlled trial. J Clin Psychopharmacol. 2015;35:43–50. This study resulted clinical implications for treatment with an atypical antidepressant in heavy drinkers who have genetic predisposition to AUD, and highlighted Mirtazapine's properties and mechanisms of action supporting the effects observed.
Martinotti G, Di NM, De Vita O, Hatzigiakoumis DS, Guglielmo R, Santucci B, et al. Low-dose topiramate in alcohol dependence: a single-blind, placebo-controlled study. J Clin Psychopharmacol. 2014;34:709–15. This trial is notable for using low doses of the anticonvulsant to minimize its common adverse effects for alcohol relapse prevention and to increase adherance to treatment. Specific outcomes discussed include effects on cravings and on behavioral symptoms of anxiety, hostility, and obsessive-compulsiveness.
Wackernah RC, Minnick MJ, Clapp P. Alcohol use disorder: pathophysiology, effects, and pharmacologic options for treatment. Subst Abuse Rehabil. 2014;5:1–12. In addition to covering FDA approved medications for AUD this article extensively reviews trials of non-approved medications for AUD and questions whether any one agent alone or a combination regimen may effectively treat individiuals with AUD.
Herrold AA, Kletzel SL, Harton BC, Chambers RA, Jordan N, Pape TL. Transcranial magnetic stimulation: potential treatment for co-occurring alcohol, traumatic brain injury and posttraumatic stress disorders. Neural Regen Res. 2014;9:1712–30. This review article explores the potential utility of transcranial magnetic stimulation (TMS) as a novel neurotherapeutic to treat co-occurring AUD, mTBI and posttraumatic stress disorder. A neurobiological framework is put forth to explain how TMS could be used as a potential therapy.
den Uyl TE, Gladwin TE, Wiers RW. Transcranial direct current stimulation, implicit alcohol associations and craving. Biol Psychol. 2015;105:37–42.
Klauss J, Penido Pinheiro LC, Silva Merlo BL, de Almeida Correia SG, Fregni F, Nitsche MA, et al. A randomized controlled trial of targeted prefrontal cortex modulation with tDCS in patients with alcohol dependence. Int J Neuropsychopharmacol. 2014;17:1793–803.
Kang EK, Kim DY, Paik NJ. Transcranial direct current stimulation of the left prefrontal cortex improves attention in patients with traumatic brain injury: a pilot study. J Rehabil Med. 2012;44:346–50.
Acknowledgments
This manuscript was made possible by funding from the Department of Veterans Affairs, Office of Research and Development, Rehabilitation Research & Development, Career Development Award, Level II RX000949-01A2 to Amy Herrold and the National Institute on Disability and Rehabilitation Research, Merit Switzer Research Fellowship Award H133F13001 to Amy Herrold. Dr. Sander received support from Grant Nos. H133B090023, H133A070043, and H133A120020 from the National Institute on Disability, Independent Living, and Rehabilitation Research (NIDILRR). NIDILRR is a Center within the Administration for Community Living (ACL), Department of Health and Human Services (HHS). The contents of this publication do not necessarily represent the policy of NIDILRR, ACL, HHS, and you should not assume endorsement by the Federal Government. The authors wish to thank Dr. Sandra Kletzel for a review of the manuscript.
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
All authors declare no conflicts of interest.
Human and Animal Rights and Informed Consent
This review article is not the result of any studies with human or animal subjects.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Dual Diagnosis
Rights and permissions
About this article

Cite this article
Herrold, A.A., Sander, A.M., Wilson, K.V. et al. Dual Diagnosis of Traumatic Brain Injury and Alcohol Use Disorder: Characterizing Clinical and Neurobiological Underpinnings. Curr Addict Rep 2, 273–284 (2015). https://doi.org/10.1007/s40429-015-0078-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40429-015-0078-3