
Abstract
Background
Members of the melanoma-associated antigen-A (MAGE-A) subfamily are overexpressed in many cancers and can drive cancer progression, metastasis, and therapeutic recurrence.
Objective
This study is the first comprehensive meta-analysis evaluating the prognostic utility of MAGE-A members in different cancers.
Methods
A systematic literature search was conducted in PubMed, Google Scholar, Science Direct, and Web of Science. The pooled hazard ratios with 95% confidence intervals were estimated to evaluate the prognostic significance of MAGE-A expression in various cancers.
Results
In total, 44 eligible studies consisting of 7428 patients from 11 countries were analysed. Univariate and multivariate analysis for overall survival, progression-free survival, and disease-free survival showed a significant association between high MAGE-A expression and various cancers (P < 0.00001). Additionally, subgroup analysis demonstrated that high MAGE-A expression was significantly associated with poor prognosis for lung, gastrointestinal, breast, and ovarian cancer in both univariate and multivariate analysis for overall survival.
Conclusion
Overexpression of MAGE-A subfamily members is linked to poor prognosis in multiple cancers. Therefore, it could serve as a potential prognostic marker of poor prognosis in cancers.
Similar content being viewed by others

Members of the melanoma-associated antigen-A (MAGE-A) subfamily are silent in normal tissues and overexpressed in many cancers and can drive cancer progression, metastasis, and therapeutic recurrence. |
MAGE-A expression profiling can be used as a prognostic indicator in cancer. |
This is the first meta-analysis to describe the utility of MAGE-A expression as a prognostic indicator in cancer. |
High MAGE-A expression is significantly associated with poor survival outcomes in lung, gastrointestinal, breast, and ovarian cancer. |
1 Introduction
Although the mortality of cancer has declined over time, it remains a significant public health problem globally. According to GLOBOCAN, cancer accounted for 18.1 million cases and 9.6 million deaths globally in 2018 [1]. Despite advances and improvements in the diagnosis and prognosis of cancer, there has been no significant improvement in patient survival. Lack of sensitive, specific, and reliable markers for early diagnosis, prognosis, and therapy selection have been attributed to the reduced survival rate in cancer [2]. Thus, developing a molecular marker for early diagnosis and prognosis of cancer is necessary to improve the clinical management of cancer. Molecular abnormalities (genetic and epigenetic dysregulation) plays a very important role in malignant transformation and can provide vital clinical information about cancer progression. Therefore, screening for these molecular abnormalities may be clinically useful as diagnostic and prognostic markers for cancer.
Cancer-testis antigens represent a large family of tumor antigen proteins showing restricted expression in germ cells. The abnormal expression of these antigen proteins is commonly observed in a variety of malignancies [3]. Melanoma-associated antigen-A (MAGE-A) was the subfamily of tumor-associated antigen first identified by Van der Bruggen et al. [4] in 1991 while investigating tumor antigens in melanoma cells. These antigens are recognized by the cytotoxic T lymphocytes to induce a robust immune response (T-cell reactivity) against developing cancer cells [4]. Based on chromosomal location and expression, human MAGE family members are broadly divided into type I and type II. Currently, over 60 MAGE family members have been identified. MAGE family members consisting of a highly conserved MAGE homology domain (MHD) comprising ~ 170 amino acids. Type 1 MAGE family (MAGE-A, B, and C subfamily) are highly expressed in numerous cancers with little or no expression in normal adult tissue. MAGE-D, E, F, G, H, L, and Necdin genes belong to the type II MAGE family and are expressed in a variety of tissue types. Type II MAGE loss is reported to affect neurodevelopmental functions, resulting in defective cognition, behavior, and development. The MHD of the MAGE-A family contribute to the bulk of the protein and show 60–80% conservation among the various family members. Despite the high sequence and structural similarities, the individual members of MAGE-A show distinct functions, which may be attributed to structural dynamism because of conformational changes [5].
The human genome consists of 11 annotated MAGE-As located at Xq28 and is completely silent in normal tissue except in male germ cell and placenta. The MAGE-A family plays a pivotal role in spermatogenesis and embryonic development. Expression of MAGE-A in testis or placenta suggests its potential role in germ cell development [5]. The detection of MAGE-A protein in early developmental stages of the central nervous system and peripheral nerves suggests its involvement in neuronal development [6]. MAGE-A is strongly associated with a malignant phenotype in breast cancer, bladder cancer, melanoma, oral cancer, lung cancer, and colorectal cancer [7,8,9,10,11,12]. High expression of MAGE-A genes is associated with poor survival outcomes in breast cancer, lung cancer, and gastric cancer [13,14,15]. Abnormal expression of MAGE-A is linked to epigenetic dysregulation in multiple cancer conditions [16]. Very interestingly, abnormal MAGE-A expression is more commonly detected in cancer cells that are malignant with invasive and metastatic capacity. Patients with cancer and abnormal expression of MAGE-A have a poor prognosis. Because of the tumor-specific expression and its role in immune evasion, MAGE-A has been extensively investigated as a target for immunotherapy [17]. Although the role of the MAGE-A family in the tumor is well-established, their role in normal cells remains elusive. Nevertheless, the value of MAGE-A expression as a prognostic marker in various tumors is yet to be established. In this meta-analysis, we systematically analyzed the prognostic value of MAGE-A expression in different cancers.
2 Materials and Methods
2.1 Ethics Statement
This meta-analysis was performed as per the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines and did not require ethical clearance [18].
2.2 Literature Search Strategy
We conducted a systematic literature search of the PubMed, Google Scholar, ScienceDirect, and Web of Science electronic databases to identify potential research articles to investigate associations between MAGE-A expression and cancer prognosis. The literature search was performed using medical subject headings (MeSH) and non-MeSH keywords: (“Melanoma associated antigen-A” OR “MAGE-A” OR “MAGE-A1” OR “MAGE-A2” OR “MAGE-A3” OR “MAGE-A4” OR “MAGE-A5” OR “MAGE-A6” OR “MAGE-A8” OR “MAGE-A9” OR “MAGE-A10” OR “MAGE-A11” OR “MAGE-A12”) AND (“cancer” OR “tumor” OR “neoplasm” OR “carcinoma”) AND “prognosis” up to 3 May 2020.
2.3 Inclusion and Exclusion Criteria
The following criteria were used to include the studies for meta-analysis: (1) clinical studies that investigated MAGE-A expression in various cancers and including histology information, (2) clinical studies reporting hazard ratios (HRs) with corresponding 95% confidence intervals (CIs) and P values for univariate and multivariate analysis, and (3) clinical studies published in English only.
The following exclusion criteria were applied: (1) reviews, case reports, abstracts, clinical trials, conference abstracts, book chapters, meta-analyses, and retracted studies; (2) studies that did not perform survival analysis; (3) studies without independent survival data for MAGE-A; (4) studies with insufficient data to calculate the standard error and perform statistical analysis; (5) studies that did not report HR or provide sufficient data to calculate HR; (6) studies not published in English; and (7) studies of only cell lines or animal models.
2.4 Data Extraction
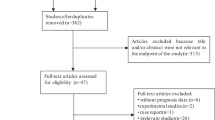
Screening and selection of relevant research articles were performed independently by two researchers (MP and PVJ) using the inclusion and exclusion criteria. Where disagreements occurred, the research articles were further screened independently by a third reviewer (SPK). Figure 1 describes this screening process. The information extracted from research papers for pooled analysis included author details, year of publication, origin of sample, sample size, sample type, cancer types, technique used for detection of MAGE-A expression, survival information (overall survival [OS], progression-free survival [PFS], and disease-free survival [DFS]) and HRs with corresponding 95% CI and P value (Table 1).

Flow chart summarizing the screening process for selection of eligible studies
2.5 Data Synthesis and Quality Assessment
The data synthesis and quality assessment was performed independently by two reviewers (MP and PVJ). The association between MAGE-A expression and cancer prognosis was investigated using Review Manager version 5.3 (The Cochrane Collaboration) and Meta-Essentials version 1.4 [19]. The relationship between MAGE-A expression and cancer prognosis was calculated using HRs with corresponding 95% CIs. The effect of the study heterogeneity on data synthesis was assessed using Cochrane’s Q test, and Higgin’s I2 test (I2 < 25% indicates no heterogeneity, I2 = 25–50% indicates moderate heterogeneity, I2 > 50% indicates high heterogeneity). Accordingly, a random-effects model and a fixed-effects model were selected to pool data with significant and non-significant heterogeneity, respectively [20]. Subgroup analysis was performed to evaluate the association between MAGE-A expression and five different cancer types. Begg’s funnel plot and Egger’s bias indicator test were implemented to identify any potential publication bias [21, 22]. A P < 0.05 was considered statistically significant.
2.6 GRADE and Statistical Analysis
Two researchers (MP and PVJ) evaluated the quality of the evidence using the GRADE (Grading of Recommendations Assessment, Development, and Evaluation) criteria. The impact of evidence on indirectness, imprecision, inconsistency, publication bias, and size effect were assessed using GRADE. The quality of evidence was rated as high, moderate, low, or very low according to Cochrane Training criteria (Cochrane Training) [23] and with reference to a study published by Creemers et al. [24] (Table 2).
A pan-cancer analysis was performed to identify the prognostic significance of the individual members of the MAGE-A gene in various cancers using a Kaplan–Meier (KM) (https://kmplot.com/analysis/) plotter. From the KM survival plots, the HRs, 95% CIs, and log-rank P values were obtained and computed. The data were analysed as per the default parameters. A P < 0.05 was considered statistically significant.
3 Results
3.1 Study Selection and Features
The literature collected up to 3 May 2020 identified 2917 potential studies. Further screening excluded 2807 studies as being either duplicate studies, abstracts, reviews, book chapters or articles written in non-English languages (Fig. 1). Screening of 110 articles identified 44 eligible studies consisting of data from 7428 patients from 11 countries with sample sizes ranging from 42 to 552 participants. Among the 44 selected studies, 22 were from China, five from Germany, four from South Korea, three each from the USA and Japan, and one each from Denmark, England, Switzerland, Brazil, Croatia, and Canada. The shortlisted studies used immunohistochemistry (IHC) and quantitative real-time polymerase chain reaction (qRT-PCR) to measure MAGE-A expression and used formalin-fixed paraffin-embedded (FFPE) tissues (n = 32), cancer tissue samples (n = 8), blood (n = 4), bone marrow (n = 1), and peritoneal wash fluid (n = 1) as the sample source (Table 1). The MAGE-A gene members and their functions in various cancers are shown in Fig. 1 and Table 1 in the ESM. The prognostic utility of individual MAGE-A gene members at the RNA and protein level is shown in Table 2 in the ESM.
3.2 Association between MAGE-A Expression and Overall Survival
The association between MAGE-A expression and OS was evaluated using both univariate and multivariate analysis. The univariate analysis included 25 studies consisting of 3450 patients, and the multivariate analysis included 33 studies with 5427 patients. Both univariate (HR 2.36 [95% CI 2.00–2.78], Z score 10.24; P < 0.00001) and multivariate analysis (HR 1.82 [95% CI 1.52–2.18], Z score 6.48; P < 0.00001) showed a significant association between MAGE-A expression and cancer (Fig. 2a, b). Significant heterogeneity was also observed in both univariate (I2 = 53%) and multivariate (I2 = 65%) analysis and was overcome by implementation of the random-effects model.

Forest plot showing the association between MAGE-A expression in cancer and overall survival: a univariate analysis and b multivariate analysis. CI confidence interval, IV inverse variance, MAGE-A melanoma-associated antigen-A, SE standard error
3.3 Association between MAGE-A Expression and Disease-Free Survival
The univariate analysis included seven studies consisting of data from 1276 patients, and the multivariate analysis included eight studies with 1682 patients. MAGE-A expression was significantly associated with DFS in the univariate analysis (HR 1.81 [95% CI 1.37–2.41], Z score 4.13; P = 0.00001) (Fig. 3a), and the multivariate analysis showed a significant association between MAGE-A expression and cancer (HR 1.56 [95% CI 1.22–2.00], Z score 3.57; P = 0.0004) (Fig. 3b). Univariate analysis (I2 = 64%) and multivariate analysis (I2 = 64%) showed high heterogeneity; therefore, a random-effects model was applied for both.

Forest plot showing the association between MAGE-A expression in cancer and disease-free survival: a univariate analysis and b multivariate analysis. CI confidence interval, IV inverse variance, MAGE-A melanoma-associated antigen-A, SE standard error
3.4 Association between MAGE-A Expression and Progression-Free Survival
Data from three studies with a sample size of 1118 patients were collected and analyzed for univariate analysis. Multivariate analysis included two studies consisting of 586 patients. Univariate analysis (HR 4.21 [95% CI 2.50–7.09], Z score 5.40; P < 0.00001) (Fig. 4a) showed a significant association between MAGE-A expression and cancer and no heterogeneity (I2 = 0%); multivariate analysis (HR 3.48 [95% CI 1.74–6.98], Z score 3.52; P = 0.0004) (Fig. 4b) showed a significant association between MAGE-A expression and cancer. No heterogeneity was observed for univariate (I2 = 0%) and multivariate analysis (I2 = 0%); therefore, a fixed-effect model was applied for both.

Forest plot showing the association between MAGE-A expression in cancer and progression-free survival: a univariate analysis and b multivariate analysis. CI confidence interval, IV inverse variance, MAGE-A melanoma-associated antigen-A, SE standard error
3.5 Subgroup Analysis
Univariate analysis was performed in 23 studies for five different cancer types: two studies in breast cancer, six in lung cancer, five in head and neck squamous cell carcinoma (HNSCC), eight in gastrointestinal cancer, and two in ovarian cancer. All five different cancer types showed a significant association between MAGE-A expression and cancer: lung (HR 2.64 [95% CI 1.82–3.83], Z score 5.09; P < 0.00001), breast cancer (HR 2.84 [95% CI 1.82–4.44], Z score 4.61; P < 0.00001), HNSCC (HR 2.94 [95% CI 1.78–4.85], Z score 4.23; P < 0.00001), gastrointestinal cancer (HR 2.20 [95% CI 1.67–2.90], Z score 5.64; P < 0.00001), and ovarian cancer (HR 2.47 [95% CI 1.66–3.68], Z score 4.48; P < 0.00001). Figure 5 shows the overall effect for univariate OS (HR 2.45 [95% CI 2.08–2.89], Z score 10.72; P < 0.00001).

Forest plot for subgroup analysis. Forest plot for effect of MAGE-A on cancer types on overall survival of univariate analysis. CI confidence interval, IV inverse variance, MAGE-A melanoma-associated antigen-A, SE standard error
Multivariate analysis was performed in 30 studies for five cancer types: breast cancer (n = 2), lung cancer (n = 7), HNSCC (n = 7), gastrointestinal cancer (n = 11), and ovarian cancer (n = 3). MAGE-A expression was significantly linked with lung cancer (HR 2.41 [95% CI 1.71–3.39], Z score 5.03; P < 0.00001), gastrointestinal cancer (HR 1.77 [95% CI 1.44–2.19], Z score 5.32; P < 0.00001), ovarian cancer (HR 1.62 [95% CI 1.14–2.31], Z score 2.69; P = 0.007), and breast cancer (HR 3.29 [95% CI 1.07–10.17], Z score 2.07; P = 0.04). However, MAGE-A expression was not significantly associated with HNSCC (HR 1.49 [95% CI 0.81–2.72], Z score 1.28; P = 0.20). Figure 6 shows the overall effect for multivariate OS (HR 1.90 [95% CI 1.59–2.28], Z score 6.98; P < 0.00001).

Forest plot for subgroup analysis. Forest plot for effect of MAGE-A on cancer types on overall survival of multivariate analysis. CI confidence interval, HNSCC head and neck cancer, IV inverse variance, MAGE-A melanoma-associated antigen-A, SE standard error
3.6 Publication Bias
The potential for publication bias was eliminated using Egger’s test and Begg’s funnel plot. Egger’s test and Begg & Mazumdar’s rank correlation test were non-significant both for univariate for OS (t = 0.85, z = 0.71; P = 0.238) and DFS (t = 1.06, z = 1.34; P = 0.091) (Fig. 7b) and for multivariate for OS (t = -0.47, z = 0.03; P = 0.489) and DFS (t = 1.19, z = 1.59; P = 0.056) (Fig. 7c). However, there was a potential publication bias for univariate analysis for PFS (t = 6.69, z = 2.44; P = 0.007) and for multivariate analysis for PFS (t = 2.95, z = 2.04; P = 0.021) (Fig. 7b,c). The combined result for both univariate and multivariate analysis for OS, as shown in Fig. 7a, indicates a lack of publication bias (t = -0.21, z = 0.42; P = 0.338).

Begg’s funnel plot for publication bias test: a publications in overall survival (both univariate and multivariate), b publications in univariate analysis for overall survival, disease-free survival, and progression-free survival; c publications in multivariate analysis for overall survival, disease-free survival, and progression-free survival. The x-axis is ln (HR), and the y-axis is the standard error of ln (HR). The horizontal line represents the overall estimated ln (HR). The two diagonal lines indicate the pseudo 95% confidence limits of the effect estimate. CES combined effect size, HR hazard ratio, ln (HR) (natural) log-transformed HR
3.7 Prognostic Values of MAGE-A Gene Members in Various Cancers
The KM plotter database was used to compute the prognostic significance of individual MAGE-A gene members. Among the 12 MAGE-A family members analyzed for OS, MAGE-A1, -A2, -A4, -A9, -A10, and -A12 were significant in HNSCC and kidney renal clear cell carcinoma (p < 0.05), whereas MAGE-A3 was significant in HNSCC and MAGE-A6 was significant in kidney renal clear cell carcinoma. MAGE-A1, -A3, -A4, -A9, -A10, and -A12 were significant in liver hepatocellular carcinoma and lung squamous cell carcinoma, whereas MAGE-A8, -A6, and -A11 were significant in liver hepatocellular carcinoma and MAGE-A2 was significant in lung squamous cell carcinoma. MAGE-A2, -A4, -A9, -A11, and -A12 were significant in ovarian cancer and pancreatic ductal adenocarcinoma, whereas MAGE-A10 was significant in ovarian and MAGE-A3 was significant in pancreatic ductal adenocarcinoma. Data for MAGE-A5 were not generated in the KM plotter. MAGE-A8 and -A9 were significant in lung adenocarcinoma, pheochromocytoma, and paraganglioma, whereas MAGE-A1 and -A3 were significant in lung adenocarcinoma and MAGE-A11 in pheochromocytoma and paraganglioma. MAGE-A2 and -A12 were significant in bladder cancer and breast cancer, whereas MAGE-A1 and -A8 were significant in bladder cancer and MAGE-A3, -A4, and -A9 were significant in breast cancer. MAGE-A2, -A9, and -A11 were significant in cervical squamous cell carcinoma and stomach adenocarcinoma, whereas MAGE-A3 and -A12 were significant in stomach adenocarcinoma. MAGE-A1, -A9, and -A12 were significant in sarcoma and thyroid cancer, whereas MAGE-A3 and -A10 were significant in sarcoma and MAGE-A4 in thyroid carcinoma. MAGE-A6 and -A9 were significant in thymoma and esophageal adenocarcinoma, whereas MAGE-A1 and -A8 were significant in esophageal adenocarcinoma and MAGE-A2, -A10, and -A11 were significant in thymoma. MAGE-A6 and -A10 were significant in kidney renal papillary cell carcinoma (Table 3 in the ESM).
4 Discussion
Despite advances in the early diagnosis, prognosis, and treatment of cancer, it remains a key public health problem around the world, suggesting the need for reliable biomarkers for early diagnostic and theranostic applications. Several clinical and molecular investigations have identified gene signatures with prognostic significance in cancer. However, many fail to be implemented in clinical setup because of (1) low sensitivity in detecting cancer in asymptomatic patients, (2) low tissue specificity, and (3) low prognostic value [2]. The association between abnormal expression of MAGE-A members and cancer is now well-established. Thus, measuring MAGE-A members either at the RNA or the protein level has potential for use as a diagnostic and prognostic cancer marker [83]. MAGE-A members are pro-tumorigenic in nature and are reported to affect key biological characteristics of cancer cells, such as growth, proliferation, migration, invasion, metastasis, and chemoresistance (Table 1 in the ESM). We assessed the prognostic value of MAGE-A gene expression because their expression is restricted to testis and placenta in normal adult tissue, but they are abnormally expressed in numerous cancerous tissues. Given the high tumor-specific expression, MAGE-A level has emerged as a potential prognostic marker and therapeutic target in cancers [83].
MAGE family members play important roles in both normal development and tumor development and progression. Although initially discovered as an antigen expressed by cancer cells, current studies indicate it has a prominent role in cancer, and it is being explored as a target for immunotherapy [84]. Many patients with cancer show overexpression of the MAGE family of proteins [83]. Success as a candidate for cancer immunotherapy has been sparse, so many studies are currently focusing on investigating the regulation and biological function of the MAGE family of genes in cancer [5]. Cancers overexpressing MAGE were more aggressive and showed the worst clinical outcomes. Various functional studies have indicated that some of the MAGE genes have non-overlapping oncogenic functions [83]. Thus, developing therapeutic targets against the MAGE family may be an attractive approach in clinical management of cancers.
The biological function of MAGE-A family members is not known, but reports suggest it can act as a master regulator of E3 RING ubiquitin ligase by enhancing their activity [71, 85]. Aberrant regulation of E3 RING ubiquitin ligases by MAGE members has been reported as contributing to tumorigenesis. Recently, MAGE-A11 has been shown to induce ubiquitination of PCF11, which results in 3′ UTR shortening of downstream tumor suppressor proteins and oncogenes, which ultimately results in increased tumorigenesis [86]. MAGE-A1 has been shown to regulate transcription by interacting with Ski interacting protein (SKIP) and recruiting HDAC1 to inhibit transcription [70]. In cancer cells, MAGE-A members show abnormal expression, leading to the acquisition of tumor-promoting properties such as tumor growth, proliferation, migration, and invasion with concomitant inhibition to apoptosis [5]. The abnormal activation of MAGE family genes is now attributed to epigenetic dysregulation such as DNA hypomethylation, defective histone modifications, and nucleosome occupancy [16]. DNA hypomethylation has been shown to induce aberrant expression of MAGE-A genes and is associated with poor survival outcomes in laryngeal squamous cell carcinoma and esophageal squamous cell carcinoma [87, 88]. By acting as a transcriptional regulator, MAGE participates in a variety of pro-tumorigenic functions. MAGE-activated KAP1 functions as a transcriptional repressor by promoting histone deacetylation and H3-K9 methylation and heterochromatinization. MAGE-A2, -A3, and -A6 are reported to bind to the coiled-coil domain of TRIM28/KAP1 ubiquitin ligases. In prostate cancer, MAGE-A activation increases androgen receptor (AR) activity, promoting cancer progression (Table 1 in the ESM). MAGE-C2, a member of the MAGE subfamily, participates in double-stranded DNA repair pathways via phosphorylation of TRIM28/KAP1, facilitating interactions between TRIM28/KAP1 and ATM [89].
MAGE-A subfamily members prevent p53 activity via multiple mechanisms. The MAGE-A-mediated activation of KAP1 is reported to repress p53 by its degradation [75]. Proteasomal-dependent p53 degradation is enhanced by MAGE-A via enhancement of the ubiquitin ligase activity of TRIM28/KAP1 [85]. Further, MAGE-A directly interacts with p53, blocking the binding of p53 to its target genes. Knockdown of MAGE-A is reported to enhance p53 levels and its recruitment to target gene promoters, enhancing the expression of p53 targets. MAGE-A binds to the DNA-binding domain of p53, repressing its transcription [90]. It also inhibits apoptosis through suppression of p53-mediated Bax expression and upregulation of survivin through p53-dependent and -independent mechanisms in multiple myeloma cells [91]. Thus, MAGE-A can act as an oncogene by inhibiting apoptosis of cancer cells. p53 activity is also downregulated by MAGE-As and by inhibiting its acetylation via HDAC3 recruitment [74]. MAGE-A3 and -A6 is reported to bring down the level of 5′ AMP-activated protein kinase (AMPK) proteins, leading to significant decreases in autophagy and activation of mammalian target of rapamycin (mTOR) signaling pathways [79]. MAGE-A11 promotes prostate cancer by activation of ARs by binding to its N-terminal FXXLF motif [81]. Epidermal growth factor-mediated phosphorylation and ubiquitination of MAGE-A11 can enhance AR activity [92]. MAGE-A11 is also a known stabilizer of hypoxia-inducible factor (HIF)-1α and thus may play a role in tumor survival [93]. The cancer stem-like cells show expression of MAGE-A2, -A3, -A4, -A6, and -A12, suggesting their essential role in the maintenance of stemness [94].
Overexpression and knockdown studies using cell lines and xenograft models have demonstrated the oncogenic potential of MAGE-As in cancer. Overexpression of MAGE-A3 has been shown to enhance the invasive potential of thyroid cancer cells [78]. Transformation of fibroblast- and anchorage-independent growth of cells was reported for MAGE-A3 and -A6 [79]. These studies suggest the oncogenic functions of MAGE-A family members. However, further studies are required for other members of the MAGE-A subfamily. MAGE-A3 and -A6 bring about degradation of p53 and AMPKα1 via activation of TRIM28, leading to loss of autophagy and mTORC1 hyperactivation, which may result in loss of growth control and induction of tumor growth [79]. MAGE-A is reported to induce proliferation of melanoma cells directly by phosphorylation of c-JUN or via the ERK-MAPK pathway [72]. Recently, MAGE-A3 overexpression has been shown to induce proliferation and migration of cervical cancer cells by modulating the EMT and Wnt signaling pathways. Therefore, these studies suggest that MAGE-A genes can induce proliferation and facilitate metastasis of cancer cells [95]. Expression of MAGE-A genes has been shown to be associated with resistance to tumor necrosis factor (TNF)-α-mediated cytotoxicity in cervical cancer cells [96]. MAGE-A overexpression is also attributed to chemoresistance. For example, MAGE-A2 confers chemoresistance in breast cancer cells by localizing to the nucleus and preventing the transactivation of p53-responsive genes, which are involved in cell cycle arrest and apoptosis in response to tamoxifen. Further, MAGE-A2 can form a complex with estrogen receptor (ER)-α, either directly or via ER cofactors, and enhance its transcriptional activity [77]. MAGE-A-mediated suppression of p53 can suppress the expression of pro-apoptotic proteins such as BIM and p21Cip1 in multiple myeloma [97]. Interestingly, MAGE-A3 can interact with long noncoding RNA (LINC01234) and microRNA (microRNA-31-5p) to facilitate proliferation and chemoresistance in hepatocellular carcinoma [98]. These findings suggest that expression of MAGE-A is associated with proliferation, inhibition of apoptosis, and chemoresistance in cancer cells. MAGE-A proteins can also inhibit autophagy and favor anabolic reactions, facilitating synthesis of macromolecules in cancer cells by downregulating AMPK through MAGE-A3/6-TRIM28 ubiquitination complex [79].
In the present meta-analysis, we collected and pooled data from 44 eligible studies with 7428 patients from 11 countries. MAGE-A overexpression in tumor tissue was positively correlated with poor clinical outcomes and recurrence risk. For instance, MAGE-A family members (A1, A3, A6, A9, and A10) are associated with the worst clinical outcomes, with poor survival rates in lung, breast, and ovarian cancer. However, their association with survival outcomes varied between cancers. For instance, Kim et al. [56] and Han et al. [55] found no association between OS and high MAGE-A expression in gastric cancer and non-Hodgkin lymphoma, respectively. As per our meta-analysis, lung, gastrointestinal, breast, and ovarian cancer showed poor OS in both univariate and multivariate analysis, whereas HNSCC showed poor OS in only univariate analysis. Additionally, pan-cancer analysis of the individual MAGE-A family members was analyzed using the KM plotter to estimate prognostic significance. The overexpression of MAGE-A2, -A3, -A4, -A9, and -A12 showed prognostic association in breast cancer, whereas MAGE-A2, -A4, -A9, -A10, -A11, and -A12 showed prognostic association with ovarian cancer; MAGE-A1, -A2, -A3, -A4, -A8, -A9, -A10, and -A12 showed association with lung cancer. and MAGE-A1, -A2, -A3, -A4, -A6, -A8, -A9, -A10, -A11, and -A12 showed prognostic significance with gastrointestinal cancers (pancreatic ductal adenocarcinoma and hepatocellular carcinoma).Thus, our meta-analysis results recommend the use of MAGE-A members as a marker for survival outcome in various cancers.
Our study has certain limitations that must be considered while interpreting the results of the study. First, study selection bias is possible, as all studies published in non-English languages were excluded from the analysis. Studies analyzing the expression of MAGE-A and another biomarker were also excluded. Expression of MAGE-A in various cancer samples was detected using two different techniques: IHC and qRT-PCR, which might have led to bias in the sensitivity of MAGE-A detection. Another limitation of our study is that all 12 MAGE-A family members were analyzed together. We could not perform the analysis on individual members of the MAGE-A gene family because either studies/data were lacking or incomplete or only a few were available. Among all the studies included in this meta-analysis, 22 studies were from China, 5 studies from Germany, 4 studies from South Korea, 3 studies from the United States of America and Japan each, and 1 study each from Denmark, England, Switzerland, Brazil, Croatia and Canada, which might have introduced a geographical bias. We have also found a potential publication bias for univariate PFS (t = 6.69, z = 2.44; P = 0.007) and multivariate PFS (t = 2.95, z = 2.04; P = 0.021). The random-effects and fixed-effects models were implemented appropriately to reduce heterogeneity bias.
5 Conclusion
MAGE-A is a cancer-testis antigen, the abnormal expression of which is linked to poor clinical outcomes in multiple cancers. To the best of our knowledge, this is the first comprehensive meta-analysis describing the prognostic utility of MAGE-A overexpression in various cancers. Our findings indicate a significant association between MAGE-A expression and OS, DFS, and PFS. Our study suggests and supports the measuring of MAGE-A levels for prognostic applications in various human malignancies.
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. https://doi.org/10.3322/caac.21492.
Diamandis EP. The failure of protein cancer biomarkers to reach the clinic: why, and what can be done to address the problem? BMC Med. 2012;10(1):87. https://doi.org/10.1186/1741-7015-10-87.
Cheng YH, Wong EWP, Cheng CY. Cancer/testis (CT) antigens, carcinogenesis and spermatogenesis. Spermatogenesis. 2011;1(3):209–20. https://doi.org/10.4161/spmg.1.3.17990.
van der Bruggen P, Traversari C, Chomez P, Lurquin C, De Plaen E, Van den Eynde B, et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science. 1991;254(5038):1643–7. https://doi.org/10.1126/science.1840703.
Lee AK, Potts PR. A comprehensive guide to the MAGE family of ubiquitin ligases. J Mol Biol. 2017;429(8):1114–42. https://doi.org/10.1016/j.jmb.2017.03.005.
Gjerstorff MF, Harkness L, Kassem M, Frandsen U, Ditzel HJ, Nielsen O, et al. Distinct GAGE and MAGE-A expression during early human development indicate specific roles in lineage differentiation. Hum Reprod. 2008;23(10):2194–201. https://doi.org/10.1093/humrep/den262.
Hou S, Sang M, Geng C, Liu W, Lü W, Xu Y, et al. Expressions of MAGE-A9 and MAGE-A11 in breast cancer and their expression mechanism. Arch Med Res. 2014;45(1):44–51. https://doi.org/10.1016/j.arcmed.2013.10.005.
Mengus C, Schultz-Thater E, Coulot J, Kastelan Z, Goluza E, Coric M, et al. MAGE-A10 cancer/testis antigen is highly expressed in high-grade non-muscle-invasive bladder carcinomas. Int J Cancer. 2013;132(10):2459–63. https://doi.org/10.1002/ijc.27914.
Brasseur F, Rimoldi D, Lienard D, Lethe B, Carrel S, Arient F, et al. Expression of mage genes in primary and metastatic cutaneous melanoma. Int J Cancer. 1995;63(3):375–80. https://doi.org/10.1002/ijc.2910630313.
Brisam M, Rauthe S, Hartmann S, Linz C, Brands RC, Kübler AC, et al. Expression of MAGE-A1-A12 subgroups in the invasive tumor front and tumor center in oral squamous cell carcinoma. Oncol Rep. 2016;35(4):1979–86. https://doi.org/10.3892/or.2016.4600.
Sienel W, Varwerk C, Linder A, Kaiser D, Teschner M, Delire M, et al. Melanoma associated antigen (MAGE)-A3 expression in Stages I and II non-small cell lung cancer: results of a multi-center study. Eur J Cardiothorac Surg. 2004;25(1):131–4. https://doi.org/10.1016/j.ejcts.2003.09.015.
Shantha Kumara H, Grieco MJ, Caballero OL, Su T, Ahmed A, Ritter E, et al. MAGE-A3 is highly expressed in a subset of colorectal cancer patients. Cancer Immun. 2012;12(1):16.
Ayyoub M, Scarlata CM, Hamai AH, Pignon P, Valmori D. Expression of MAGE-A3/6 in primary breast cancer is associated with hormone receptor negative status, high histologic grade, and poor survival. J Immunother. 2014;37(2):73–6. https://doi.org/10.1097/CJI.0000000000000013.
Yoshida N, Abe H, Ohkuri T, Wakita D, Sato M, Noguchi D, et al. Expression of the MAGE-A4 and NY-ESO-1 cancer-testis antigens and T cell infiltration in non-small cell lung carcinoma and their prognostic significance. Int J Oncol. 2006;28(5):1089–98. https://doi.org/10.3892/ijo.28.5.1089.
Ogata K, Aihara R, Mochiki E, Ogawa A, Yanai M, Toyomasu Y, et al. Clinical significance of melanoma antigen-encoding gene-1 (MAGE-1) expression and its correlation with poor prognosis in differentiated advanced gastric cancer. Ann Surg Oncol. 2011;18(4):1195–203. https://doi.org/10.1245/s10434-010-1399-z.
Lian Y, Meng L, Ding P, Sang M. Epigenetic regulation of MAGE family in human cancer progression-DNA methylation, histone modification, and non-coding RNAs. Clin Epigenet. 2018;10(1):115. https://doi.org/10.1186/s13148-018-0550-8.
Gjerstorff MF, Andersen MH, Ditzel HJ. Oncogenic cancer/testis antigens: prime candidates for immunotherapy. Oncotarget. 2015;6(18):15772–87. https://doi.org/10.18632/oncotarget.4694.
Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. Ann Intern Med. 2009;151(4):264–9. https://doi.org/10.7326/0003-4819-151-4-200908180-00135.
Suurmond R, van Rhee H, Hak T. Introduction, comparison, and validation of meta-essentials: a free and simple tool for meta-analysis. Res Synth Methods. 2017;8(4):537–53. https://doi.org/10.1002/jrsm.1260.
DerSimonian R, Laird N. Meta-analysis in clinical trials. Control Clin Trials. 1986;7:177–88. https://doi.org/10.1016/0197-2456(86)90046-2.
Begg CB, Mazumdar M. Operating characteristics of a rank correlation test for publication bias. Biometrics. 1994. https://doi.org/10.2307/2533446.
Egger M, Smith GD, Schneider M, Minder C. Bias in meta-analysis detected by a simple, graphical test. Br Med J. 1997;315(7109):629–34. https://doi.org/10.1136/bmj.315.7109.629.
Cochrane Training. Introduction to GRADE. https://training.cochrane.org/introduction-grade. Accessed 12 Dec 2019.
Creemers H, Grupstra H, Nollet F, van den Berg LH, Beelen A. Prognostic factors for the course of functional status of patients with ALS: a systematic review. J Neurol. 2015;262(6):1407–23. https://doi.org/10.1007/s00415-014-7564-8.
Balafoutas D, Zur Hausen A, Mayer S, Hirschfeld M, Jaeger M, Denschlag D, et al. Cancer testis antigens and NY-BR-1 expression in primary breast cancer: prognostic and therapeutic implications. BMC Cancer. 2013;13(1):271. https://doi.org/10.1186/1471-2407-13-271.
Chen X, Wang L, Liu J, Huang L, Yang L, Gao Q, et al. Expression and prognostic relevance of MAGE-A3 and MAGE-C2 in non-small cell lung cancer. Oncol Lett. 2017;13(3):1609–18. https://doi.org/10.3892/ol.2017.5665.
Dyrskjøt L, Zieger K, Lildal TK, Reinert T, Gruselle O, Coche T, et al. Expression of MAGE-A3, NY-ESO-1, LAGE-1 and PRAME in urothelial carcinoma. Br J Cancer. 2012;107(1):116–22. https://doi.org/10.1038/bjc.2012.215.
Gu L, Sang M, Yin D, Liu F, Wu Y, Liu S, et al. MAGE-A gene expression in peripheral blood serves as a poor prognostic marker for patients with lung cancer. Thorac Cancer. 2018;9(4):431–8. https://doi.org/10.1111/1759-7714.12571.
Gu X, Fu M, Ge Z, Zhan F, Ding Y, Ni H, et al. High expression of MAGE-A9 correlates with unfavorable survival in hepatocellular carcinoma. Sci Rep. 2014. https://doi.org/10.1038/srep06625.
Gure AO, Chua R, Williamson B, Gonen M, Ferrera CA, Gnjatic S, et al. Cancer-testis genes are coordinately expressed and are markers of poor outcome in non-small cell lung cancer. Clin Cancer Res. 2005;11(22):8055–62. https://doi.org/10.1158/1078-0432.CCR-05-1203.
Han L, Jiang B, Wu H, Zhang S, Lu X. Expression and prognostic value of MAGE-A9 in laryngeal squamous cell carcinoma. Int J Clin Exp Pathol. 2014;7(10):6734–42.
Lian Y, Sang M, Gu L, Liu F, Yin D, Liu S, et al. MAGE-A family is involved in gastric cancer progression and indicates poor prognosis of gastric cancer patients. Pathol Res Pract. 2017;213(8):943–8. https://doi.org/10.1016/j.prp.2017.05.007.
Liu S, Sang M, Xu Y, Gu L, Liu F, Shan B. Expression of MAGE-A1, -A9, -A11 in laryngeal squamous cell carcinoma and their prognostic significance: a retrospective clinical study. Acta Otolaryngol. 2016;136(5):506–13. https://doi.org/10.3109/00016489.2015.1126856.
Mecklenburg I, Sienel W, Schmid S, Passlick B, Kufer P. A threshold of systemic MAGE-A gene expression predicting survival in resected non-small cell lung cancer. Clin Cancer Res. 2017;23(5):1213–9. https://doi.org/10.1158/1078-0432.CCR-16-0557.
Noh ST, Lee HS, Lim SJ, Kim SW, Chang HK, Oh J, et al. MAGE-A1–6 expression in patients with head and neck squamous cell carcinoma: impact on clinical patterns and oncologic outcomes. Int J Clin Oncol. 2016;21(5):875–82. https://doi.org/10.1007/s10147-016-0989-6.
Sang M, Gu L, Liu F, Lian Y, Yin D, Fan X, et al. Prognostic significance of MAGE-A11 in esophageal squamous cell carcinoma and identification of related genes based on DNA microarray. Arch Med Res. 2016;47(3):151–61. https://doi.org/10.1016/j.arcmed.2016.06.001.
Sang M, Wu X, Fan X, Lian Y, Sang M. MAGE-A family serves as poor prognostic markers and potential therapeutic targets for epithelial ovarian cancer patients: a retrospective clinical study. Gynecol Endocrinol. 2017;33(6):480–4. https://doi.org/10.1016/j.arcmed.2016.06.001.
Sang M, Wu X, Fan X, Sang M, Zhou X, Zhou N. Multiple MAGE-A genes as surveillance marker for the detection of circulating tumor cells in patients with ovarian cancer. Biomarkers. 2014;19(1):34–42. https://doi.org/10.3109/1354750X.2013.865275.
Ujiie H, Kato T, Lee D, Hu HP, Fujino K, Kaji M, et al. Overexpression of MAGEA2 has a prognostic significance and is a potential therapeutic target for patients with lung cancer. Int J Oncol. 2017;50(6):2154–70. https://doi.org/10.3892/ijo.2017.3984.
Wang M, Li J, Wang L, Chen X, Zhang Z, Yue D, et al. Combined cancer testis antigens enhanced prediction accuracy for prognosis of patients with hepatocellular carcinoma. Int J Clin Exp Pathol. 2015;8(4):3513–28.
Wu J, Wang J, Shen W. Identification of MAGEA12 as a prognostic outlier gene in gastric cancers. Neoplasma. 2017;64(2):238–43. https://doi.org/10.4149/neo_2017_210.
Xu X, Tang X, Lu M, Tang Q, Zhang H, Zhu H, et al. Overexpression of MAGE-A9 predicts unfavorable outcome in breast cancer. Exp Mol Pathol. 2014;97(3):579–84. https://doi.org/10.1016/j.yexmp.2014.11.001.
Xu Y, Wang C, Zhang Y, Jia L, Huang J. Overexpression of MAGE-A9 is predictive of poor prognosis in epithelial ovarian cancer. Sci Rep. 2015. https://doi.org/10.1038/srep12104.
Xylinas E, Cha EK, Khani F, Kluth LA, Rieken M, Volkmer BG, et al. Association of oncofetal protein expression with clinical outcomes in patients with urothelial carcinoma of the bladder. J Urol. 2014;191(3):830–41. https://doi.org/10.1016/j.juro.2013.08.048.
Zhai X, Xu L, Zhang S, Zhu H, Mao G, Huang J. High expression levels of MAGE-A9 are correlated with unfavorable survival in lung adenocarcinoma. Oncotarget. 2016;7(4):4871–81. https://doi.org/10.18632/oncotarget.6741.
Zhan W, Zhang Z, Zhang Y, Ma J, Wu T, Gu Y, et al. Prognostic value of MAGE-A9 expression in patients with colorectal cancer. Clin Res Hepatol Gastroenterol. 2015;40(2):239–45. https://doi.org/10.1016/j.clinre.2015.08.005.
Zhang S, Zhai X, Wang G, Feng J, Zhu H, Xu L, et al. High expression of MAGE-A9 in tumor and stromal cells of non-small cell lung cancer was correlated with patient poor survival. Int J Clin Exp Pathol. 2015;8(1):541–50.
Coombes RC, Caballero OL, Shousha S, Ghaem-Maghami S, Woodley-Barker L, Wilhelm-Benartzi CS, et al. NY-ESO-1 expression in DCIS: A new predictor of good prognosis. Oncoscience. 2017;4(3–4):33–40. https://doi.org/10.18632/oncoscience.348.
Cuffel C, Rivals JP, Zaugg Y, Salvi S, Seelentag W, Speiser DE, et al. Pattern and clinical significance of cancer-testis gene expression in head and neck squamous cell carcinoma. Int J Cancer. 2011;128(11):2625–34. https://doi.org/10.1002/ijc.25607.
Jeon CH, Kim IH, Chae HD. Prognostic value of genetic detection using CEA and MAGE in peritoneal washes with gastric carcinoma after curative resection: result of a 3-year follow-up. Medicine. 2014;93:11. https://doi.org/10.1097/MD.0000000000000083.
Kim J, Reber HA, Hines OJ, Kazanjian KK, Tran A, Ye X, et al. The clinical significance of MAGEA3 expression in pancreatic cancer. Int J Cancer. 2006;118(9):2269–75. https://doi.org/10.1002/ijc.21656.
Zamunér FT, Karia BT, de Oliveira CZ, dos Santos CR, Carvalho AL, Vettore AL. A comprehensive expression analysis of cancer testis antigens in head and neck squamous cell carcinoma revels MAGEA3/6 as a marker for recurrence. Mol Cancer Ther. 2015;14(3):828–34. https://doi.org/10.1158/1535-7163.MCT-14-0796.
Gu L, Sang M, Li J, Liu F, Wu Y, Liu S, et al. Expression and prognostic significance of MAGE-A11 and transcription factors (SP1, TFCP2 and ZEB1) in ESCC tissues. Pathol Res Pract. 2019;215(7):152446. https://doi.org/10.1016/j.prp.2019.152446.
Zhou JX, Li Y, Chen SX, Deng AM. Expression and prognostic significance of cancer-testis antigens (CTA) in intrahepatic cholagiocarcinoma. J Exp Clin Cancer Res. 2011;30:1. https://doi.org/10.1186/1756-9966-30-2.
Han MH, Eom HS, Park WS, Yun T, Park S, Kim HJ, et al. Detection of circulating lymphoma cells in patients with non-Hodgkin lymphoma using MAGE-A3 gene expression in peripheral blood. Leukemia Res. 2010;34(9):1127–31. https://doi.org/10.1016/j.leukres.2009.11.028.
Kim WH, Jung EJ, Kim HS, Spiessens B, Gruselle O, Kusuma N, et al. Expression of MAGE-A3 and/or PRAME tumor antigens in South Korean gastric cancer patients. Int J Clin Exp Pathol. 2016;9(9):9086–96.
Haier J, Owzcareck M, Guller U, Spagnoli GC, Bürger H, Senninger N, et al. Expression of MAGE-A cancer/testis antigens in esophageal squamous cell carcinomas. Anticancer Res. 2006;26(3B):2281–7.
Bergeron A, Picard V, LaRue H, Harel F, Hovington H, Lacombe L, et al. High frequency of MAGE-A4 and MAGE-A9 expression in high-risk bladder cancer. Int J Cancer. 2009;125(6):1365–71. https://doi.org/10.1002/ijc.24503.
Laban S, Giebel G, Klümper N, Schröck A, Doescher J, Spagnoli G, et al. MAGE expression in head and neck squamous cell carcinoma primary tumors, lymph node metastases and respective recurrences-implications for immunotherapy. Oncotarget. 2017;8(9):14719–35. https://doi.org/10.18632/oncotarget.14830.
Faiena I, Astrow SH, Elashoff DA, Jain R, Bot A, Chamie K, et al. Melanoma-associated antigen-A and programmed death-ligand 1 expression are associated with advanced urothelial carcinoma. Cancer Immunol Immunother. 2019;68(5):743–51. https://doi.org/10.1007/s00262-019-02316-w.
Yu Y, Huang C, Li Z, Zhao F, Zhou Y, Li J, et al. Expressions of melanoma-associated antigen A1 as a prognostic factor in Chinese patients with resectable oesophageal squamous cell carcinoma. Interact Cardiovasc Thorac Surg. 2019;29(4):510–6. https://doi.org/10.1093/icvts/ivz141.
Baba T, Shiota H, Kuroda K, Shigematsu Y, Ichiki Y, Uramoto H, et al. Clinical significance of human leukocyte antigen loss and melanoma-associated antigen 4 expression in smokers of non-small cell lung cancer patients. Int J Clin Oncol. 2013;18(6):997–1004. https://doi.org/10.1007/s10147-012-0491-8.
Sang M, Gu L, Yin D, Liu F, Lian Y, Zhang X, et al. MAGE-A family expression is correlated with poor survival of patients with lung adenocarcinoma: a retrospective clinical study based on tissue microarray. J Clin Pathol. 2017;70(6):533–40. https://doi.org/10.1136/jclinpath-2016-203718.
Tang WW, Liu ZH, Yang TX, Wang HJ, Cao XF. Upregulation of Magea4 correlates with poor prognosis in patients with early stage of esophageal squamous cell carcinoma. OncoTargets Ther. 2016;9:4289–93. https://doi.org/10.2147/OTT.S99195.
Srdelić S, Kuzmić-Prusac I, Spagnoli GC, Juretić A, Čapkun V. MAGE-A4 and MAGE-A1 Immunohistochemical Expression in High-grade Endometrial Cancer. Int J Gynecol Pathol. 2019;38(1):59–655. https://doi.org/10.1097/PGP.0000000000000470.
Lausenmeyer EM, Braun K, Breyer J, Gierth M, Denzinger S, Burger M, Voelker HU, Otto W. Strong expression of cancertestis antigens CTAG1B and MAGEA3 is correlated with unfavourable histopathological features and MAGEA3 is associated with worse progression-free survival in urothelial bladder cancer. Urol Int. 2019;102(1):77–82. https://doi.org/10.1159/000493577.
Endo M, Kanda M, Sawaki K, Shimizu D, Tanaka C, Kobayashi D, Hattori N, Hayashi M, Yamada S, Koike M, Omae K. Tissue expression of melanoma-associated antigen A6 and clinical characteristics of gastric cancer. Anticancer Res. 2019;39(11):5903–10. https://doi.org/10.21873/anticanres.13794.
Jia S, Zhang M, Li Y, Zhang L, Dai W. MAGE-A11 expression predicts patient prognosis in head and neck squamous cell carcinoma. Cancer Manag Res. 2020;12:1427.
De Smet C, De Backer O, Faraoni I, Lurquin C, Brasseur F, Boon T. The activation of human gene MAGE-1 in tumor cells is correlated with genome-wide demethylation. Proc Natl Acad Sci. 1996;93:7149–53. https://doi.org/10.1073/pnas.93.14.7149.
Laduron S, Deplus R, Zhou S, Kholmanskikh O, Godelaine D, De Smet C, et al. MAGE-A1 interacts with adaptor SKIP and the deacetylase HDAC1 to repress transcription. Nucleic Acids Res. 2004;32(14):4340–50. https://doi.org/10.1093/nar/gkh735.
Kozakova L, Vondrova L, Stejskal K, Charalabous P, Kolesar P, Lehmann AR, et al. The melanoma-associated antigen 1 (MAGEA1) protein stimulates the E3 ubiquitin-ligase activity of TRIM31 within a TRIM31-MAGEA1-NSE4 complex. Cell Cycle. 2015;14(6):920–30. https://doi.org/10.1080/15384101.2014.1000112.
Wang D, Wang J, Ding N, Li Y, Yang Y, Fang X, et al. MAGE-A1 promotes melanoma proliferation and migration through C-JUN activation. Biochem Biophys Res Commun. 2016;473(4):959–65. https://doi.org/10.1016/j.bbrc.2016.03.161.
Wischnewski F, Pantel K, Schwarzenbach H. Promoter demethylation and histone acetylation mediate gene expression of MAGE-A1, -A2, -A3, and-A12 in human cancer cells. Mol Cancer Res. 2006;4:339–49. https://doi.org/10.1158/1541-7786.MCR-05-0229.
Monte M, Simonatto M, Peche LY, Bublik DR, Gobessi S, Pierotti MA, et al. MAGE-A tumor antigens target p53 transactivation function through histone deacetylase recruitment and confer resistance to chemotherapeutic agents. Proc Natl Acad Sci. 2006;103(30):11160–5. https://doi.org/10.1073/pnas.0510834103.
Yang B, O'Herrin SM, Wu J, Reagan-Shaw S, Ma Y, Bhat KM, et al. MAGE-A, mMage-b, and MAGE-C proteins form complexes with KAP1 and suppress p53-dependent apoptosis in MAGE-positive cell lines. Cancer Res. 2007;67(20):9954–62. https://doi.org/10.1158/0008-5472.CAN-07-1478.
Marcar L, Ihrig B, Hourihan J, Bray SE, Quinlan PR, Jordan LB, et al. MAGE-A cancer/testis antigens inhibit MDM2 ubiquitylation function and promote increased levels of MDM4. PLoS ONE. 2015;10:5. https://doi.org/10.1371/journal.pone.0127713.
Wong PP, Yeoh CC, Ahmad AS, Chelala C, Gillett C, Speirs V, et al. Identification of MAGEA antigens as causal players in the development of tamoxifen-resistant breast cancer. Oncogene. 2014;33(37):4579–88. https://doi.org/10.1038/onc.2014.45.
Liu W, Cheng S, Asa SL, Ezzat S. The melanoma-associated antigen A3 mediates fibronectin-controlled cancer progression and metastasis. Cancer Res. 2008;68(19):8104–12. https://doi.org/10.1158/0008-5472.CAN-08-2132.
Pineda CT, Ramanathan S, Tacer KF, Weon JL, Potts MB, Ou YH, et al. Degradation of AMPK by a cancer-specific ubiquitin ligase. Cell. 2015;160(4):715–28. https://doi.org/10.1016/j.cell.2015.01.034.
De Smet C, Lurquin C, Lethé B, Martelange V, Boon T. DNA methylation is the primary silencing mechanism for a set of germ line-and tumor-specific genes with a CpG-rich promoter. Mol Cell Biol. 1999;19(11):7327–35.
Askew EB, Bai S, Blackwelder AJ, Wilson EM. Transcriptional synergy between melanoma antigen gene protein-A11 (MAGE-11) and p300 in androgen receptor signaling. J Biol Chem. 2010;285(28):21824–36.
Su S, Chen X, Geng J, Minges JT, Grossman G, Wilson EM. Melanoma antigen-A11 regulates substrate-specificity of Skp2-mediated protein degradation. Mol Cell Endocrinol. 2017;439:1–9. https://doi.org/10.1016/j.mce.2016.10.006.
Weon JL, Potts PR. The MAGE protein family and cancer. Curr Opin Cell Biol. 2015;37:1–8. https://doi.org/10.1016/j.ceb.2015.08.002.
Sang M, Wang L, Ding C, Zhou X, Wang B, Wang L, et al. Melanoma-associated antigen genes—an update. Cancer Lett. 2011;302(2):85–90. https://doi.org/10.1016/j.canlet.2010.10.021.
Doyle JM, Gao J, Wang J, Yang M, Potts PR. MAGE-RING protein complexes comprise a family of E3 ubiquitin ligases. Mol Cell. 2010;39(6):963–74. https://doi.org/10.1016/j.molcel.2010.08.029.
Yang SW, Li L, Connelly JP, Porter SN, Kodali K, Gan H, Park JM, Tacer KF, Tillman H, Peng J, Pruett-Miller SM. A cancer-specific ubiquitin ligase drives mRNA alternative polyadenylation by ubiquitinating the mRNA 3′ end processing complex. Mol Cell. 2020;77(6):1206–21. https://doi.org/10.1016/j.molcel.2019.12.022.
Liu S, Zhao Y, Xu Y, Sang M, Zhao R, Gu L, Shan B. The clinical significance of methylation of MAGE-A1 and-A3 promoters and expression of DNA methyltransferase in patients with laryngeal squamous cell carcinoma. Am J Otolaryng. 2020;41(1):102318. https://doi.org/10.1016/j.amjoto.2019.102318.
Gu L, Sang M, Li J, Liu F, Wu Y, Liu S, Shan B. Demethylation-mediated upregulation of melanoma-associated antigen-A11 correlates with malignant progression of esophageal squamous cell carcinoma. Digest Liver Dis. 2019;51(10):1475–82. https://doi.org/10.1016/j.dld.2019.04.018.
Bhatia N, Xiao TZ, Rosenthal KA, Siddiqui IA, Thiyagarajan S, Smart B, et al. MAGE-C2 promotes growth and tumorigenicity of melanoma cells, phosphorylation of KAP1, and DNA damage repair. J Invest Dermatol. 2013;133(3):759–67. https://doi.org/10.1038/jid.2012.355.
Marcar L, MacLaine NJ, Hupp TR, Meek DW. Mage-A cancer/testis antigens inhibit p53 function by blocking its interaction with chromatin. Cancer Res. 2010;70(24):10362–70. https://doi.org/10.1158/0008-5472.CAN-10-1341.
Nardiello T, Jungbluth AA, Mei A, DiLiberto M, Huang X, Dabrowski A, et al. MAGE-A inhibits apoptosis in proliferating myeloma cells through repression of Bax and maintenance of survivin. Clin Cancer Res. 2011;17(13):4309–19. https://doi.org/10.1158/1078-0432.CCR-10-1820.
Bai S, He B, Wilson EM. Melanoma antigen gene protein MAGE-11 regulates androgen receptor fnction by modulating the interdomain interaction. Mol Cell Biol. 2005;25(4):1238–57. https://doi.org/10.1128/MCB.25.4.1238-1257.2005.
Aprelikova O, Pandolfi S, Tackett S, Ferreira M, Salnikow K, Ward Y, et al. Melanoma antigen-11 inhibits the hypoxia-inducible factor prolyl hydroxylase 2 and activates hypoxic response. Cancer Res. 2009;69(2):616–24. https://doi.org/10.1158/0008-5472.CAN-08-0811.
Yamada R, Takahashi A, Torigoe T, Morita R, Tamura Y, Tsukahara T, et al. Preferential expression of cancer/testis genes in cancer stem-like cells: proposal of a novel sub-category, cancer/testis/stem gene. Tissue Antigens. 2013;81(6):428–34. https://doi.org/10.1111/tan.12113.
Gao X, Chen G, Cai H, Wang X, Song K, Liu L, Qiu T, He Y. Aberrantly enhanced melanoma-associated antigen (MAGE)-A3 expression facilitates cervical cancer cell proliferation and metastasis via actuating Wnt signaling pathway. Biomed Pharmacother. 2020;122:109710. https://doi.org/10.1016/j.biopha.2019.109710.
Park JH, Kong GH, Lee SW. hMAGE-A1 overexpression reduces TNF-alpha cytotoxicity in ME-180 cells. Mol Cells. 2002;14(1):122–9.
Mei AH, Tung K, Han J, Perumal D, Laganà A, Keats J, Auclair D, Chari A, Jagannath S, Parekh S, Cho HJ. MAGE-A inhibit apoptosis and promote proliferation in multiple myeloma through regulation of BIM and p21Cip1. Oncotarget. 2020;11(7):727. https://doi.org/10.18632/Foncotarget.27488.
Chen Y, Zhao H, Li H, Feng X, Tang H, Zhang J, Fu B, Qiu C. LINC01234/microRNA-31-5p/MAGEA3 axis mediates the proliferation and chemoresistance of hepatocellular carcinoma cells. Mol Ther Nucleic Acids. 2020;19:168–78. https://doi.org/10.1016/j.omtn.2019.10.035.
Nagy Á, Lánczky A, Menyhárt O, Győrffy B. Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci Rep. 2018;8(1):1–9. https://doi.org/10.1038/s41598-018-27521-y.
Acknowledgements
Open access funding provided by Manipal Academy of Higher Education, Manipal. The authors acknowledge the infrastructure support from TIFAC CORE in Pharmacogenomics, Government of India; DST-FIST, Government of India for infrastructure support. The funding source had no involvement in the conduct of the research and/or preparation of this article. We would also like to thank Pradyumna Jayaram for providing suggestions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
Manish Poojary, Padacherri Vethil Jishnu, and Shama Prasada Kabekkodu have no conflicts of interest that are directly relevant to the content of this article.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Poojary, M., Jishnu, P.V. & Kabekkodu, S.P. Prognostic Value of Melanoma-Associated Antigen-A (MAGE-A) Gene Expression in Various Human Cancers: A Systematic Review and Meta-analysis of 7428 Patients and 44 Studies. Mol Diagn Ther 24, 537–555 (2020). https://doi.org/10.1007/s40291-020-00476-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40291-020-00476-5