
Abstract
Immune checkpoint inhibitors (ICIs) have revolutionised the treatment landscape across many solid organ malignancies and form part of routine clinical practice in many tumours. As indications for monotherapy, doublet therapy and combination approaches with chemotherapy and targeted agents expand, clinicians must be aware of the wide range of possible immune-related adverse events (irAEs). Common toxicities, including rash, colitis, hepatitis and pneumonitis are well described in the literature, and have established diagnostic and management algorithms. Rarer toxicities, often with an incidence of less than 1%, are less defined. These syndromes can be poorly recognised, may take on a fulminant course and do not have established or evidence-based diagnostic and management strategies. As such, patients may experience increased morbidity, mortality and poorer outcomes, related both to these irAEs as well as how the treatment of these may affect the management of their underlying malignancy. In this review, we aim to explore the incidence, potential biomarkers, pathogenesis, diagnostic work-up and clinical sequelae of a selection of uncommon irAEs, with a focus on myocarditis, neurological and haematologic syndromes. Further prospective research is required to accurately define the incidence and pathogenesis of these conditions, with the aim of increasing clinician awareness of rare irAEs and to assist with a more personalised and mechanism-based approach to these syndromes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
With increased use of immune checkpoint inhibitors (ICIs), a wide spectrum of rare immune-related adverse events (irAEs) are emerging. |
Clinician awareness of rare irAEs is required for timely investigation and management. Delayed recognition can lead to high morbidity, mortality and poor overall outcomes for these patients. |
Further understanding of the pathophysiology of these conditions will improve selection of appropriate biomarkers to help inform targeted approaches to management. |
1 Introduction
The advent of immune checkpoint inhibitors (ICIs) has revolutionised the treatment landscape across a variety of solid tumours, with high rates of responses and durability observed. These agents exert their effect by restoring and bolstering the anti-tumour immune response and reversing mechanisms of tumour immune evasion via T cell driven mechanisms [1]. Anti-programmed-cell death 1 (PD-1), anti-programmed-cell death 1 ligand 1 (PD-L1) and anti-cytotoxic-T lymphocyte antigen 4 (CTLA-4) are amongst the most commonly used ICIs, as monotherapy or in combination. In addition, a variety of novel agents have been studied and are under development in clinical trials. As the indications for ICI monotherapy, doublet therapy and combinations with other targeted and chemotherapeutic agents expand, and with increased utilisation of these agents in earlier lines of treatment, clinicians are increasingly being faced with a wide spectrum of immune-related adverse events (irAEs). These events occur broadly due to loss of self-tolerance, with a wide array of inflammatory syndromes and involvement of a range of immune cells and mechanisms. While it is widely accepted that patients with pre-existing autoimmune conditions are at a higher risk of developing irAEs and that these toxicities occur more frequently with combination immunotherapy, there is minimal information regarding further predisposing factors for these conditions, which complicates patient selection for treatment [2, 3].
Well-recognised ICI toxicities include colitis, pneumonitis, hepatitis, endocrinopathies and skin toxicity [4]. The incidence of each toxicity varies across different tumour types and with different combinations of treatment, as demonstrated in Tables 1 and 2. Furthermore, as ICI use has become more widespread, rare toxicities have emerged that may not have manifested or were unrecognised in pivotal Phase III studies. For clinically significant irAEs, management involves withholding the ICI (temporarily or permanently) and administering corticosteroids, the formulation and dosage of which are guided by the severity of the irAE.
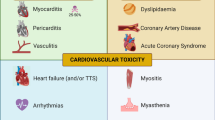
A variety of steroid-sparing immunosuppressive and immunomodulating agents have been utilised depending on the organs involved. While the majority of treatment algorithms and guidelines have been extrapolated from studies of various autoimmune diseases, emerging data suggest that the pathogenesis of irAEs differs from these conditions, and prospective studies evaluating treatments are infrequent and often limited to single-arm approaches [5]. Rarer toxicities, as demonstrated in Fig. 1, include myocarditis, neurologic and haematologic syndromes. In addition, commonly affected organ systems can also exhibit an array of inflammatory syndromes with different pathogenic mechanisms and phenotypic features. For example, the liver is commonly impacted by hepatitis but only case reports exist of sinusoidal obstruction syndrome (SOS). Although it is widely accepted that ICIs may display a wide range of off-target effects, the scope of which has not been completely defined, rarer toxicities tend to have a higher morbidity and mortality, owing both to delayed recognition and treatment and sometimes more fulminant disease course. It should be noted however, that while the incidence of fatal irAEs across a selection of Phase III clinical trials has been reported between 0.3 and 1.3%, this is lower than that of platinum doublet chemotherapy and many targeted therapies [1].

A selection of rare immune-related adverse events (irAEs) and associated frequencies. AIHA autoimmune haemolytic anaemia, GBS Guillain-Barre syndrome, HLH haemophagocytic lymphohistiocytosis, ITP idiopathic thrombocytopenia, PRCA pure red cell aplasia, SOS sinusoidal obstruction syndrome, TTP thrombotic thrombocytopenic purpura. Figure created with BioRender.com
Beyond the immediate sequelae of irAEs, clinicians must consider the impact of withholding ICIs and the administration of immunosuppressive treatments on the natural history of the underlying malignancy. It has been postulated that the development of certain irAEs may be associated with prognostic benefit in some tumour types [6,7,8,9]. While there is a growing body of observational data suggesting this, robust prospective data to validate this metric are lacking and for low frequency irAEs this is more challenging to infer. In this review, we aim to explore the incidence, potential biomarkers, pathogenesis, diagnostic work-up and clinical sequelae of a selection of uncommon irAEs, with a focus on myocarditis, neurological and haematologic syndromes. While a wide range of literature was consulted to prepare this narrative review, the authors have not undertaken a systematic review.
2 Myocarditis
Myocarditis is a rare, but potentially fatal irAE. An incidence of between 0.14 and 1.14% has been reported. Clinical features are varied and can include asymptomatic cardiac biomarker elevations (e.g., troponin and creatinine kinase [CK]), electrocardiographic (ECG) abnormalities, cardiac symptoms such as chest pain, dyspnoea, syncope and palpitations and symptoms related to a more generalised myositis including muscle pain and weakness, dysphonia, dysphagia, ptosis and diplopia. Severe cases can take a fulminant course and result in overt cardiovascular collapse [18, 19]. Given such a wide-ranging spectrum of possible presentations, recognition and diagnosis is often poor and the reported frequency likely under-represents the true incidence of this condition. In addition, a mortality rate as high as 50% has been reported, underscoring the need to define risk factors, improve diagnostic tools and develop effective management strategies [19, 20]. While combination immunotherapy approaches have been associated with higher rates of myocarditis, there are minimal data to inform further predisposing factors [2, 3]. Intrinsic factors including genetic susceptibility and gender may have a role to play, noting female predominance in ICI-related myocarditis, which is in contrast to other acute myocarditis syndromes [2]. Further patient-related cardiovascular risk factors may increase the risk of myocarditis, including obesity, hypertension, hypercholesterolaemia and smoking [3]. Interestingly, a previous history of ischaemic heart disease, atrial fibrillation and chronic kidney disease, although well-established risk factors for cardiovascular disease, are not noted to increase the risk of ICI-related myocarditis [3]. The median time to the development of myocarditis is reported to be 30 days, indicating that it tends to arise early in the treatment course [21]. Tumour-related factors may also influence the development of toxicity, either through alterations in the tumour microenvironment or through the presence of a shared antigen to specific tissues [3, 20]. It is clear from mouse models and post-mortem analyses that the pathogenesis of ICI-myocarditis is distinct, and involves immune-cell infiltrates rich in T cells and macrophages, with minimal antibody-mediated inflammation. Possible mechanisms of immune-related cardiac toxicity include the failure of peripheral tolerance and the presence of autoreactive T cells, tumour-antigen cross-reactivity and cytokine- (particularly IL-17a) mediated damage [2]. It is interesting to note that the most frequently coexisting irAEs alongside myocarditis are myositis and myasthenia gravis (both discussed below). Syndromes involving inflammation of smooth muscle have not been reported. This adds weight to the theory of a shared antigen between the tumour and striated muscle [20].
Despite our understanding of some of the mechanisms responsible for ICI-myocarditis, reliable screening strategies and biomarkers are lacking. Some clinicians advocate the use of baseline ECG, troponin and brain natriuretic peptide (BNP), which may then be repeated for the first few cycles of treatment, during which the risk of development of myocarditis is highest [22]. While these may provide useful comparator measurements for those who subsequently develop myocarditis, there is no consistent evidence to suggest that patients with baseline conduction abnormalities or elevations in troponin are at a higher risk of developing myocarditis. Moreover, while troponins are frequently elevated in cases of ICI-myocarditis, there have been reported cases of myocarditis with relatively normal troponin levels [3, 22]. Similarly, BNP, a marker of left atrial stretch and usually indicative of ventricular dysfunction, may be normal in cases of myocarditis with preserved ejection fraction, which may occur in up to 50% of cases [3, 21]. Structural imaging of cases of ICI-myocarditis has also provided varied results. There have also been reports of a Takotsubo-like syndrome of apical ballooning, calling into question the role of ICI in exacerbating cardiac stress. Cardiac magnetic resonance imaging (MRI) has often been lauded as an important component of the diagnostic workup, but 46% of cases of myocarditis may not demonstrate inflammation or abnormalities in late gadolinium enhancement, adding to the challenge of establishing a diagnostic criteria for ICI-myocarditis [3, 22]. Endomyocardial biopsy, which demonstrates T-cell and macrophage-rich infiltrates within the myocardium, is the gold-standard in diagnosis of ICI-related myocarditis [2]. However, access can be challenging due to lack of expertise and potential complications of the procedure, so is not always feasible.
Management of irAEs follows the general principles of ICI interruption, immunosuppression and supportive care. Current evidence suggests that initiation of high-dose corticosteroids or additional immunosuppression within 24 hours of development of myocarditis yields the best outcomes [5]. However, due to the rarity and sometimes minimally or atypically symptomatic nature of ICI-myocarditis, there can be delays in recognition, diagnosis and subsequent treatment [20]. The optimal duration of corticosteroid treatment is unknown but guidelines suggest patients are weaned off these medications slowly, often over a period of 4–6 weeks [5, 23]. In steroid refractory cases, a variety of alternate immunosuppressive agents and strategies have been used, including mycophenolate, tacrolimus, anti-thymocyte globulin, alemtuzumab, tocilizumab and plasmapheresis [2, 5, 19]. Although prospective data are limited, one study prospectively evaluated the combination of ruxolitinib (JAK inhibitor) and abatacept (recombinant CTLA4-analogue) in patients with ICI-induced myocarditis. Although this was a small single-arm series of 40 patients, results indicated use of this combination dramatically reduced mortality rates over time, from 50% to approximately 3% [19]. While early mouse model studies demonstrated efficacy of abatacept in the management of ICI-myocarditis, it was noted that time to onset was slow, with 10 weeks of treatment required prior to reduction of myocardial immune infiltrate [2, 19]. It was subsequently noted that JAK-STAT signalling was also upregulated in mouse models with ICI-myocarditis, leading to the rationale for ruxolitinib use. The addition of ruxolitinib has been shown to enhance abatacept activity, shortening the time of onset to hours [19]. While randomised clinical trials will provide more robust data in this space, these results are promising for the future of mechanistically directed therapies for irAEs [2, 19].
The long-term sequalae of ICI-myocarditis are not well defined. Current data suggest that in those patients who survive an episode of myocarditis, there is an increased incidence of subsequent major cardiac events and an accelerated rate of atherosclerosis [3]. Although the exact mechanism of this phenomenon is incompletely understood, it has been suggested that a heightened inflammatory state, including cell- and cytokine-mediated mechanisms, may contribute, as is the case in many autoimmune and rheumatic conditions that may also pose additional cardiovascular risk to patients [3].
3 Neurological Toxicity
Neurological irAEs are varied and have been reported at a frequency of 0.3–6% [24,25,26,27]. These may include both central (encephalitis, meningitis, demyelinating syndromes) and peripheral (peripheral neuropathies, neuromuscular disorders, myositis) syndromes [25, 28]. Interestingly, development of immune-related neurotoxicity has been associated with improved survival in several series [25, 29, 30].
3.1 Encephalitis
Encephalitis is the most frequent central neurological toxicity reported, comprising approximately 13% of neurological irAEs [25]. Mortality rates are high and range from 5 to 32% [31, 32]. Due to clinical similarities with central nervous system (CNS) malignancy, vascular events, paraneoplastic, autoimmune and infectious syndromes, it can be difficult to identify [24, 32]. Onset tends to be early, with a median onset of 8 weeks from treatment initiation. However, this is variable and cases have been reported after 6 months of treatment and even after treatment completion [1, 24]. Immune checkpoint inhibitor-encephalitis appears to be more common with the use of anti-PD-1/PD-L1 agents, and less so with anti-CTLA-4 or combination regimes [25]. Clinical features are diverse, but there is a tendency towards cortical symptoms, with impaired consciousness, disorientation, confusion and aphasia amongst the most commonly reported [32]. Paraneoplastic and autoimmune antibody presence is variable, with one study reporting only 6% of ICI-induced encephalitis syndromes being antibody positive while other reports suggest a higher presence, ranging from 37 to 58% [25, 32]. In studies that reported a higher prevalence of paraneoplastic and autoimmune antibodies, anti-Ma2 and anti-Hu antibodies were the most frequent [25]. Cerebrospinal fluid (CSF) analysis was amongst the most valuable diagnostic tests, demonstrating inflammation, with high protein levels and lymphocytic pleocytosis, as well as ruling out infectious differentials [25, 32]. While some patients demonstrate abnormal MRI findings corresponding to their clinical syndrome, this was not a reliable finding and many patients had normal imaging [24]. However, MRI is an important diagnostic tool to rule out structural and vascular aetiologies of neurological symptoms [32]. Interestingly, despite ICI interruption, and withdrawal in some cases, oncological response was maintained in patients who experienced encephalitis. As compared to paraneoplastic and autoimmune encephalitis, patients who experienced ICI-induced encephalitis tended to respond better to treatment and had better long-term outcomes, with higher rates of complete neurological recovery reported [24, 25, 32]. This finding, in addition to the varied presentations amongst ICI-induced encephalitis syndromes, suggests mechanistic differences between each inflammatory condition resulting in distinct patterns of CNS inflammation [32].
3.2 Aseptic Meningitis
Aseptic meningitis is a rare neurological irAE, which, at times, may display overlapping clinical features with encephalitis. Incidence is reported between 0.1 and 0.2%, with typical onset between 6 to 9 weeks from initiation of ICIs [31, 33]. Incidence is higher with anti-CTLA-4 agents and combination regimens [33]. Little is known about additional risk factors or pathogenesis, although pre-existing onco-neuronal antibody presence has been postulated as a possible predisposing factor [31]. Clinical features are often non-specific and include headache, fever, cognitive changes and gait disturbance, but can display a fulminant course [33]. Diagnostic work-up includes lumbar puncture, which typically demonstrates a sterile lymphocytic pleocytosis and MRI brain, which demonstrates diffuse leptomeningeal enhancement in 42% of cases [33]. At present, no biochemical- or imaging-based biomarkers to predict risk of development or clinical course of ICI-induced aseptic meningitis have been identified. As the main differentials for ICI-induced aseptic meningitis are infectious meningitis or paraneoplastic syndromes, the work-up of which usually takes time to yield results, management of these patients usually involves a combination of corticosteroids, antibiotics and antivirals to address all possibilities. In immune-mediated cases, response is typically observed within 24–48 h, but slow corticosteroid taper over 4–6 weeks may be employed to mitigate the risk of relapse [33]. Most patients experience clinical resolution without major neurological sequelae [33].
3.3 Cerebral Vasculitis
Large-vessel vasculitis and vasculitis syndromes of the nervous system have been reported with ICI use, with a preponderance for anti-PD-1 agents [34, 35]. This may be classified further into primary and secondary angiitis of the CNS [35]. Clinical syndromes may vary according to the predominant vessels involved, and range from focal syndromes (e.g., focal seizures, specific movement disorders, cerebellar signs) to more generalised neurological manifestations, with features of non-specific meningoencephalitis [27]. Onset is variable, but usually occurs within 3 months of ICI commencement [27, 34]. There are no specific autoantibodies associated with ICI-induced CNS vasculitis syndromes [27]. Due to the vague nature of presentations, differentials are wide and investigation is similar to previously described neurologic irAEs, including CSF examination and MRI brain. Cerebrospinal fluid typically demonstrates lymphocytic pleocytosis. The MRI brain may demonstrate multiple bilateral hyperintense foci of restricted diffusion. This can be associated with microhaemorrhages, leptomeningeal and cranial nerve enhancement [27]. Treatment paradigms are not defined, but once again include initiation of high-dose corticosteroids. Alternate immunosuppressive agents have been used for refractory cases, including cyclophosphamide, azathioprine and mycophenolate [27].
3.4 Neuromuscular Conditions and Myositis
Myositis is amongst the more common neurological irAEs, comprising approximately 30% of cases, acknowledging their rare occurrence [25]. Reported myositis symptoms have varied, ranging from isolated CK rises to severe myopathy. Limb-girdle weakness was most commonly reported, but neck and facial muscle involvement was seen in 29% of cases [5, 25]. When investigated, 36% of patients were found to have positive myositis-specific antibodies [25]. Electromyography (EMG) revealed a classical myopathic pattern in the majority of patients, and muscle MRI consistently demonstrated muscle oedema. Muscle biopsies often confirmed necrotising and inflammatory changes [25, 36]. Management includes corticosteroids as well as consideration of concurrent intravenous immunoglobulin (IVIg), or plasmapheresis [25, 36]. Clinical symptoms and CK can be used to track response to treatment [36]. Although most patients respond to treatment, mortality was still reported at 17%, with most patients dying due to respiratory failure or sudden cardiac events [25].
As an immunological privileged site, ophthalmic irAEs are rare, with an incidence of 1–3% [37, 38]. Ocular myositis is a rare subtype of myositis syndrome in which there is isolated involvement of extraocular muscles. There is significant clinical overlap with ocular myasthenic syndromes (discussed below), with symptoms including diplopia, ptosis and complex ophthalmoplegias, and as such, diagnosis can be challenging [36]. Pain on eye movements, absence of fatiguability and a myopathic EMG pattern are important differentiators between the conditions. Presence or absence of antibodies, either myositis-specific or acetylcholine-receptor (AChR) antibodies, are less reliable in differentiating these conditions, as both may be seronegative, or display overlapping syndromes [36]. Management is similar to other myositis syndromes (discussed above).
Myasthenic syndromes are often severe, with bulbar and respiratory involvement seen in a majority of patients, and infrequent isolated ocular myasthenia [25]. These were observed more commonly with anti-PD-1/PD-L1 agents than anti-CTLA-4 or combination therapy. Co-existing myopathy is common [20]. Interestingly, 58% of patients demonstrated positive AChR antibodies but anti-muscle–specific kinase (MusK) was not demonstrated [25]. Although most patients responded well to corticosteroid treatment without significant relapse issues, mortality remained high (28%) and was mostly in the setting of respiratory failure [25].
It should be noted that myositis, myasthenia gravis and myocarditis are three frequently co-occurring irAEs. This has raised the possibility of a common antigenic trigger, which potentially shares a similar structure to tumour antigen [3, 4, 20, 25]. Moreover, similarities between myositis and myasthenic syndromes, including clinical symptoms, antibody positivity and mechanism of mortality, have led to suggestions that these toxicities represent a spectrum of the same condition [25].
While T cells are typically thought of as being the main drivers of irAEs, autoantibody positivity is relatively frequent in neurologic irAEs, sometimes existing even prior to ICI treatment [39]. While the pathogenic significance of these antibodies is unclear, it does raise the question about whether antibodies can be used to identify those who may be more prone to developing neurologic irAEs, and could perhaps be used as a biomarker [25]. Small numbers and retrospective analyses limit definitive statements regarding this question, and further prospective work is required in this space. Moreover, in patients with antibody positivity prior to treatment, the question of whether ICI is in fact unmasking a paraneoplastic phenomenon should be considered. Clinical differences between paraneoplastic syndromes and irAEs suggest alternate mechanisms, but the potential interaction between these two factors warrants further examination [25].
3.5 Guillain-Barre Peripheral Neuropathy
Immune checkpoint inhibitor-induced Guillain–Barre syndrome (GBS) is a rare peripheral neurological irAE with a reported incidence of 0.1–0.3% [26, 40]. Cases are observed with both single-agent and combination ICI use with no predilection for either therapeutic approach. Mortality rates are high (11–27%), with 10% of patients dying from respiratory muscle paralysis [25, 26]. The pathogenesis of ICI-mediated GBS is unclear, but there have been suggestions of shared antigens between gangliosides found in melanoma cells and Schwann cells. Interestingly, both melanocytes and Schwann cells originate from the neural crest, supporting a common epitope theory [26]. Clinical features include progressive weakness, hyporeflexia/areflexia and paraesthesia [26]. While typical GBS syndromes are often associated with an infectious trigger leading to immune-mediated demyelination of peripheral nerves, ICI-induced GBS exhibits some key clinical and diagnostic differences. While there is a male predominance in both syndromes, this appears more pronounced in ICI-related GBS, with 79% of ICI-induced GBS cases occurring in men in one study [26]. Median onset in one study was 8.2 weeks from ICI onset, which is later than infectious cases [26]. Cerebrospinal fluid examination is inflammatory and reveals elevated protein levels, with typical albumin-cytological differentiation observed in 63.9% of patients [26]. Electromyography has revealed different subtypes of GBS syndromes in association with ICIs, acute inflammatory demyelinating polyneuropathy, acute motor axonal neuropathy and Miller-Fisher syndrome [25, 26]. Treatment paradigms are undefined but involve a combination of corticosteroids, IVIg and plasmapheresis. Interestingly, corticosteroids are not routinely used in cases of idiopathic GBS, but are sometimes effective in ICI-induced cases [25]. Approximately 70% of patients demonstrated partial or complete recovery of symptoms after treatment with these strategies [25, 26].
4 Haematological Toxicity
Haematological irAEs are rare, with an incidence under 5% [41,42,43,44,45]. They remain poorly defined in the literature and recognition can be challenging, in part due to the increasing frequency of combining ICIs with myelosuppressive chemotherapy. Haematological irAEs predominantly affect single haematopoietic lineages, less commonly involving multiple lineages; however, a wide range of conditions has been reported. These include red cell syndromes (haemolysis and pure red cell aplasia [PRCA]), neutropenia, haemophagocytic lymphohistocytosis (HLH), acquired thrombotic or bleeding disorders such as haemophilia A and thrombotic thrombocytopenic purpura (TTP) [43, 46,47,48,49]. Onset of haematological irAEs tends to be early in the treatment course (median 6 to 8 weeks), but varies widely [43, 46, 50,51,52]. Prompt recognition of haematological irAEs and involvement of a haematologist in the diagnostic work-up and management is important due to the high proportion of patients developing life threatening toxicity with reported mortality rates of up to 14% [41].
4.1 Cytopenias
Idiopathic thrombocytopenic purpura (ITP), autoimmune haemolytic anaemia (AIHA), and neutropenia are the most commonly observed ICI-related cytopenias with an incidence of around 0.2% [41, 43]. Pure red cell aplasia and aplastic anaemia, which results in pancytopenia, have also been reported although are considered very rare [53,54,55,56]. Mechanisms underlying the risk of ICI-induced cytopenias are unclear and likely to be multifactorial, but are believed to commonly involve the production of autoantibodies. Underlying haematological malignancies and autoimmune conditions of any type may increase the risk of developing immune-related cytopenias, particularly AIHA, but no other pre-disposing factors have been clearly identified to date [42, 50, 57,58,59,60]. The diagnostic work-up of ICI-induced cytopenias should include a thorough screen for alternative causes including history, examination and reviewing concurrent medications. Investigations will depend upon the affected cell lineage(s) but include evaluation of the peripheral blood smear, direct anti-globulin test (DAT), haemolysis markers, viral serology and low threshold for bone-marrow examination. It is noteworthy that the incidence of DAT positivity amongst cases of ICI-associated AIHA may be lower than in primary AIHA and diagnosis should not be excluded on this basis [58]. Management of these conditions mirrors that of their non-iatrogenic counterparts. In addition to withholding the ICI, supportive measures such as growth factors and transfusion, are commonly required. High-dose corticosteroids are usually recommended, with between 50 to 70% of cases being steroid responsive [43, 61]. Hence, many patients will require additional immunosuppression for steroid-refractory events and will typically respond to similar therapies given for non-ICI–driven cytopenias, with agents such as IVIg, rituximab, mycophenolate mofetil, azathioprine and cyclosporine A. A significant portion of ICI-induced cytopenias are life-threatening with 44–89% of affected patients developing grade 4 events and up to 5% mortality [42, 46, 50]. One series demonstrated the incidence of grade 4 neutropenia to be 0.14% [46]. While resolution occurs for the majority of patients, recovery is often protracted (months) with the exception of aplastic anaemia, which tends to be permanent [42, 46, 58].
4.2 Haemophagocytic Lymphohistiocytosis
Immune checkpoint inhibitor-associated HLH is a rare and potentially fatal toxicity characterised by T cell and macrophage hyper-activation, subsequent uncontrolled release of pro-inflammatory cytokines resulting in tissue injury and multi-organ dysfunction. In some series, HLH is more commonly associated with PD-1 than CTLA-4 inhibitors, although this is not universal and its incidence has been reported with both therapeutic classes [48, 51, 59]. It appears to affect men more than women. Timing of onset from ICI initiation is widely variable with reported averages across case series ranging from 6 to 18 weeks. Patients typically present with fever, organomegaly, pancytopenia, significantly elevated ferritin and low fibrinogen. Features of haemophagocytosis may be evident on bone marrow aspiration, but not universally [62, 63]. The diagnosis of HLH is often delayed due to lack of clinician awareness of the condition as well as overlapping differential diagnoses with similar symptoms and laboratory abnormalities. In addition, HLH may be driven by co-existing confounding factors such as malignancy or infection, which may be challenging to distinguish. Pre-defined criteria may be used to establish the diagnosis (HLH-2004 and HScore) [64]. Treatment begins with cessation of the ICI and management algorithms align with the treatment of non-ICI–related HLH with prompt initiation of high-dose corticosteroids. One unique therapeutic advance is that early addition of the IL-6 antagonist tocilizumab has proven useful in the management of ICI-associated HLH [65, 66]. As such, international guidelines for the management of drug-induced HLH (including ICI) recommend tocilizumab in addition to corticosteroids and consideration of etoposide if no response [64]. Early recognition and aggressive management of ICI-associated HLH is critical, due to associated mortality of up to one-fourth of patients [48, 51, 52].
4.3 Acquired Bleeding Disorders
Acquired haemophilia A due to ICI therapy has been reported with PD-1, PD-L1 and CTLA-4 inhibitors [49, 67,68,69]. This is a rare bleeding disorder associated with auto-antibodies to factor VIII. Onset tends to be early and has been reported between 8 to 14 weeks after commencing ICIs [68, 69]. Patients generally present with bleeding, which can be severe and life-threatening. Bleeding manifestations tend to be into skin, muscle, mucous membranes, gastrointestinal and urinary tract. Laboratory analyses demonstrate prolonged APTT, not correcting with mixing of normal plasma and evidence of an acquired factor VIII inhibitor [49, 68]. Patients have been successfully treated with corticosteroids, recombinant factor VII, rituximab and cyclophosphamide [69,70,71]. Thrombotic thrombocytopenic purpura (TTP) is a rare blood disorder associated with high mortality characterised by microangiopathic haemolytic anaemia (MAHA), thrombocytopenia and microvascular occlusion [72]. The phenotype is derived from deficiency of ADAMTS-13, a metalloprotease, which cleaves large von Willebrand factor (vWF) multimers. Thrombotic thrombocytopenic purpura may be acquired due to inhibitory autoantibodies, which can develop in the context of infection, malignancy, pregnancy or medication and has been reported with ICI [73]. The historical clinical pentad includes thrombocytopenia, MAHA, neurological dysfunction and fever; however, in practice this presentation is rare [72]. The diagnosis of acquired TTP is made by confirming absence of ADAMTS-13 function and the presence of autoantibodies [74]. Despite a very high mortality and many patients dying due to TTP, it has been successfully treated with plasmapheresis, corticosteroids, rituximab and caplacizumab [47, 73, 75,76,77].
Although the risk of serious or life-threatening AEs is high, haematological irAEs will commonly resolve with immunosuppression and withholding ICIs. Cessation of ICIs has significant implications for the underlying malignancy hence the question of whether patients can be safely re-challenged is important. There is a relatively high rate of recurrence of haematological irAEs upon re-exposure, between 14 to 43%, therefore careful consideration is warranted when deciding to recommence ICI therapy [42, 43, 50, 58, 61]. If recommencing ICI therapy, vigilance for recurrence of haematological irAEs is important and warrants close monitoring.
5 Other Rare Toxicities
5.1 Hepatobiliary: Sinusoidal Obstruction Syndrome
Sinusoidal obstruction syndrome (SOS) is an obliterative venulitis characterised by fibrotic damage of small hepatic vessels [78]. It is typically associated with allogenic haematopoietic stem cell transplants, but cases have been reported in patients with colorectal cancer with liver metastases post-cytoreductive surgery as well as with the drug gemtuzumab ozogamicin [79]. Five cases of SOS have been reported in patients with metastatic melanoma who received ICIs without additional recognised risk factors for this condition [78,79,80]. Predisposing factors and pathogenesis have not been elucidated. Clinical features include painful hepatomegaly, fluid overload and deranged liver function tests with raised bilirubin. Imaging may demonstrate a pattern of heterogenous enhancement in the portal phase and MRI can be helpful in assessment [79]. Invasively measured portal pressures may be elevated [78, 79]. Histological examination reveals non-fibrous portal areas with sinusoidal dilation and perisinusoidal fibrosis with lobular veins occluded with fibrous tissue [78]. Cases have been managed with a combination of ICI cessation, corticosteroids and mycophenolate mofetil. In addition, supportive medications, mainly for the symptoms of fluid overload, have been used [78, 79]. The role of defibrotide in ICI-induced SOS is unclear [79]. These strategies resulted in clinical and biochemical improvements. However, one patient died due to disease progression 6 months after cessation of ICI [78].
5.2 Hepatobiliary: Cholangitis
Immune checkpoint inhibitor-induced cholangitis is a rare irAE with a reported incidence of 0.05–0.7% [81]. Onset is variable, with cases being reported from 1 week to 18 months post-treatment initiation [81,82,83]. Clinical features are similar to other cholangitis-type syndromes, including right upper quadrant pain, fever and jaundice. Patients are usually symptomatic, although rare cases of asymptomatic bilirubin and cholestatic liver enzyme elevations have been described [82]. Important differential diagnoses to rule out include other autoimmune conditions such as primary sclerosing cholangitis, primary biliary cirrhosis and IgG4 disease, as well as infectious and malignant aetiologies [81]. Diagnostic work-up involves a thorough autoimmune panel, including anti-nuclear antibodies, anti-mitochondrial antibodies and anti-nuclear cytoplasmic antibodies, as well as IgG4 levels. Diagnostic imaging using CT or MRI to visualise the liver and biliary tree is also useful [81, 82]. In cases involving large, extrahepatic bile ducts, imaging findings can include segmental or diffuse non-obstructive dilation and stenosis of the biliary duct lumen. There may be enhancement, hypertrophy and irregularity of bile duct walls. Adjacent structures such as the gallbladder, can also be affected demonstrating wall thickening and oedema [81]. Histopathological findings obtained via endoscopic retrograde cholangiopancreatography vary according to the proportion of small and large duct involvement, but features may include CD8 lymphocyte-rich periportal inflammation, damage to small bile ducts, ductular reactions and loss of bile ducts. If extra-hepatic bile duct involvement predominates, additional features may include intraepithelial lymphocytic infiltration and diffuse non-ventricular fibrosis of ducts [81]. Management involves corticosteroids, immunosuppressive agents and immunomodulatory agents titrated to effect. It should be noted that infliximab is avoided as an immunosuppressive agent in ICI-induced cholangitis due to the risk of precipitating liver failure [84]. Ursodeoxycholic acid is also used as a supportive medication [82]. When compared to irAE hepatitis, it has been noted that ICI-induced cholangitis and biliary syndromes tend to be less corticosteroid responsive, complicating rechallenge and subsequent therapies [81, 83].
5.3 Endocrinopathies: Hypoparathyroidism
Immune checkpoint inhibitor-induced endocrinopathies are common, affecting 10% of patients managed with ICIs, with thyroiditis and hypophysitis being amongst the most commonly reported syndromes [85, 86]. Immune checkpoint inhibitor-related hypoparathyroidism, is a rare entity with a reported incidence of 0.18–0.28% of all endocrinopathies, with most data available from case reports [86]. Time to onset is variable, with cases being reported from 6 weeks post-ICI initiation to 18 months following cessation [82, 86, 87]. Clinical features range from asymptomatic biochemical abnormalities to severe symptomatic hypocalcaemia requiring hospitalisation. Symptoms of hypocalcaemia include perioral paraesthesia and numbness, neuromuscular irritability with positive Chvostek’s and Trousseau’s signs and ECG abnormalities including prolonged QTc [82]. Biochemical features include hypocalcaemia with a low parathyroid hormone (PTH) level. Normal magnesium levels are required to exclude a diagnosis of pseudohypoparathyroidism. Furthermore, it is important to explore a past history of neck surgery or irradiation and familial cases of hypocalcaemia to exclude more commonly recognised aetiologies of hypoparathyroidism [86]. In addition, investigation of other glandular involvement is important to rule out autoimmune polyglandular syndrome 1, of which hypoparathyroidism is the most common manifestation [82, 86]. While the pathophysiology is incompletely understood, potential mechanisms include antibody-mediated activation of the calcium-sensing receptor (CaSR) resulting in reduced PTH secretion or T cell-mediated damage directly to the parathyroid gland [86]. Due to its rare occurrence, predictive biomarkers have not been established for this syndrome [85]. Management is focussed on long-term calcium and vitamin D replacement, and as is the case with most ICI-induced endocrinopathies, corticosteroid and immunosuppressive therapy is not usually required [82, 86]. Recombinant PTH use is not recommended [86]. Immune checkpoint inhibitor therapy can be continued alongside supportive therapy [82].
5.4 Genitourinary: Non-Infectious Cystitis
Immune checkpoint inhibitor-related non-infectious cystitis is a rare irAE described in case reports [88, 89]. Cases appear more common with anti-PD-1/PD-L1 use and this has been postulated to be related to PD-L1 expression in normal urothelium [82, 89]. Clinical symptoms include dysuria, frequency, urgency and haematuria. Symptoms do not respond to empiric antibiotic courses. Urine microscopy and culture reveals sterile urine, leucocytosis and erythrocytosis. Cytology is negative for malignant cells [82, 88, 89]. In some cases, renal tract imaging has demonstrated hydronephrosis without an obstructing lesion. Cystoscopy demonstrates macroscopic inflammation [89]. Biopsies demonstrate non-specific inflammation with interstitial oedema and epithelial abscission. Immunohistochemistry reveals CD8-positive T cell restricted intracellular antigen (TIA)-positive cytotoxic T cells, which are key differentiators between immune-mediated and other types of inflammation [82, 88, 89]. The presence of these cells has led to hypotheses that ICI-induced cystitis occurs as a result of TIA cells targeting an unknown urothelial antigenic trigger [82]. Symptoms respond quickly to corticosteroids, but can relapse if weaned quickly. Urinary parameters normalise within 1 to 4 weeks [89]. There are no data on the use of additional immunosuppressive agents [82, 88, 89].
5.5 Generalised Oedema Syndromes
Immune checkpoint inhibitor-induced generalised oedema syndromes are a recently reported rare irAE with an incidence of 0.19% in a single retrospective study of 20 patients [80]. Despite its low incidence, symptoms are severe (50% grade 4 toxicity) and mortality rate is high (20%). Median onset from ICI initiation was 14.5 weeks [80]. Serositis and endothelial dysfunction are potential pathophysiologic mechanisms, which initiate these syndromes. Weight gain due to fluid retention is the most common symptom, and other clinical features are related to the serosal surface involved. This may include pleural and pericardial effusions, ascites and peripheral oedema [80]. There was considerable phenotypic variation within this small study, with 9 (45%) patients demonstrating other associated syndromes, including 3 (15%) patients with SOS, 4 (20%) patients with capillary leak syndrome and 2 (10%) patients with subcutaneous autoimmune syndrome [80]. Patients were managed with varying doses and formulations of corticosteroids, with 67% of patients improving and only 2 (10%) patients requiring additional immunosuppression [80]. Additional supportive management, including diuretics, drainage and syndrome-directed therapy (e.g., ursodeoxycholic acid and defibrotide for SOS) have also been used to manage these syndromes. Data are lacking regarding this rare irAE and it is unclear whether the varied oedema syndromes within this small group of patients represent distinct pathophysiological mechanisms or a spectrum of the same condition.
6 Conclusions
Immune checkpoint inhibitors used as monotherapy, doublet therapy and in combination with other oncological agents, are established within the treatment paradigms of various solid organ malignancies. Ever-expanding use of these agents has drawn attention to a wide spectrum of possible toxicities mediated by an unregulated immune system. While common toxicities, including rash, colitis, pneumonitis and hepatitis, are frequently reported in the literature, rarer toxicities, including myocarditis, neurological and haematological toxicities are less well understood, and tend to have a high morbidity and mortality [4]. Sequalae of these events can be prolonged and have a significant impact on patients’ quality of life and may impact choice of therapies and overall oncological outcomes [4]. The decision regarding whether to rechallenge ICI following a toxicity is nuanced, balancing risk of irAE relapse with disease progression, and greater understanding of the pathophysiology and likely clinical consequences of this requires further elucidation. Our review of rare irAEs demonstrates key differences between ICI-induced conditions and their idiopathic autoimmune counterparts. We have also noted a paucity of high-quality prospective data necessary to establish the true incidence, risk factors and potential biomarkers of these conditions and whether they predict outcomes. Given the practical difficulties in conducting large-scale clinical trials with a focus on irAEs, further study of real-world registry data, such as is collected in the Side Effect Registry in Immuno-Oncology (SERIO) database may be helpful [90]. A greater awareness of the wide-ranging clinical effects of ICIs as well as an understanding of the underlying pathogenesis and mechanisms of immune dysfunction will allow more personalised assessment of risks and benefits in selection of therapy as well as inform a more tailored treatment approach. This has the potential to mitigate the requirement for long courses of corticosteroids and lower the risk of relapse, thus improving the quality of life of patients whose longevity has been significantly improved with ICIs.
References
Martins F, Sofiya L, Sykiotis GP, Lamine F, Maillard M, Fraga M, et al. Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat Rev Clin Oncol. 2019;16(9):563–80. https://doi.org/10.1038/s41571-019-0218-0.
Wei SC, Meijers WC, Axelrod ML, Anang NAS, Screever EM, Wescott EC, et al. A genetic mouse model recapitulates immune checkpoint inhibitor-associated myocarditis and supports a mechanism-based therapeutic intervention. Cancer Discov. 2021;11(3):614–25. https://doi.org/10.1158/2159-8290.CD-20-0856.
Patel RP, Parikh R, Gunturu KS, Tariq RZ, Dani SS, Ganatra S, et al. Cardiotoxicity of immune checkpoint inhibitors. Curr Oncol Rep. 2021;23(7):79. https://doi.org/10.1007/s11912-021-01070-6.
Wang DY, Salem J-E, Cohen JV, Chandra S, Menzer C, Ye F, et al. Fatal toxic effects associated with immune checkpoint inhibitors: a systematic review and meta-analysis. JAMA Oncol. 2018;4(12):1721. https://doi.org/10.1001/jamaoncol.2018.3923.
Haanen J, Obeid M, Spain L, Carbonnel F, Wang Y, Robert C, et al. Management of toxicities from immunotherapy: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann Oncol. 2022;33(12):1217–38. https://doi.org/10.1016/j.annonc.2022.10.001.
Haratani K, Hayashi H, Chiba Y, Kudo K, Yonesaka K, Kato R, et al. Association of immune-related adverse events with nivolumab efficacy in non–small-cell lung cancer. JAMA Oncol. 2018;4(3):374. https://doi.org/10.1001/jamaoncol.2017.2925.
Wang Y, Zou J, Li Y, Jiao X, Wang Y, Zhuo N, et al. Serological biomarkers predict immune-related adverse events and clinical benefit in patients with advanced gastrointestinal cancers. Front Immunol. 2022;13: 987568. https://doi.org/10.3389/fimmu.2022.987568.
Yasuda Y, Fujiwara R, Oguchi T, Komai Y, Numao N, Yamamoto S, et al. Prognostic significance of immune-related adverse events in metastatic renal cell carcinoma patients treated with immune-checkpoint-inhibitors. Cancer Diagn Progn. 2023;3(3):327–33. https://doi.org/10.21873/cdp.10219.
Thompson LL, Nadelmann ER, Blum AE, Yoon J, Polyakov NJ, Kagan RD, et al. Patterns and prognostic significance of cutaneous immune-related adverse events in non-small cell lung cancer. Eur J Cancer. 2021;147:13–6. https://doi.org/10.1016/j.ejca.2021.01.022.
Larkin J, Chiarion-Sileni V, Gonzalez R, Grob J-J, Rutkowski P, Lao CD, et al. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2019;381(16):1535–46. https://doi.org/10.1056/NEJMoa1910836.
Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. 2016;375(19):1823–33. https://doi.org/10.1056/NEJMoa1606774.
Motzer RJ, Tannir NM, McDermott DF, Aren Frontera O, Melichar B, Choueiri TK, et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N Engl J Med. 2018;378(14):1277–90. https://doi.org/10.1056/NEJMoa1712126.
Burtness B, Harrington KJ, Greil R, Soulieres D, Tahara M, De Castro G, et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): a randomised, open-label, phase 3 study. Lancet. 2019;394(10212):1915–28. https://doi.org/10.1016/S0140-6736(19)32591-7.
Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim T-Y, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med. 2020;382(20):1894–905. https://doi.org/10.1056/NEJMoa1915745.
Oh D-Y, Ruth He A, Qin S, Chen L-T, Okusaka T, Vogel A, et al. Durvalumab plus gemcitabine and cisplatin in advanced biliary tract cancer. NEJM Evid. 2022. https://doi.org/10.1056/EVIDoa2200015.
Janjigian YY, Shitara K, Moehler M, Garrido M, Salman P, Shen L, et al. First-line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): a randomised, open-label, phase 3 trial. Lancet. 2021;398(10294):27–40. https://doi.org/10.1016/S0140-6736(21)00797-2.
Powles T, Park SH, Voog E, Caserta C, Valderrama BP, Gurney H, et al. Avelumab maintenance therapy for advanced or metastatic urothelial carcinoma. N Engl J Med. 2020;383(13):1218–30. https://doi.org/10.1056/NEJMoa2002788.
Baas P, Scherpereel A, Nowak AK, Fujimoto N, Peters S, Tsao AS, et al. First-line nivolumab plus ipilimumab in unresectable malignant pleural mesothelioma (CheckMate 743): a multicentre, randomised, open-label, phase 3 trial. Lancet. 2021;397(10272):375–86. https://doi.org/10.1016/S0140-6736(20)32714-8.
Salem J-E, Bretagne M, Abbar B, Leonard-Louis S, Ederhy S, Redheuil A, et al. Abatacept/ruxolitinib and screening for concomitant respiratory muscle failure to mitigate fatality of immune-checkpoint inhibitor myocarditis. Cancer Discov. 2023;13(5):1100–15. https://doi.org/10.1158/2159-8290.CD-22-1180.
Jiménez-Alejandre R, Ruiz-Fernández I, Martín P. Pathophysiology of immune checkpoint inhibitor-induced myocarditis. Cancers. 2022;14(18):4494. https://doi.org/10.3390/cancers14184494.
Hu J-R, Florido R, Lipson EJ, Naidoo J, Ardehali R, Tocchetti CG, et al. Cardiovascular toxicities associated with immune checkpoint inhibitors. Cardiovasc Res. 2019;115(5):854–68. https://doi.org/10.1093/cvr/cvz026.
Lyon AR, Yousaf N, Battisti NML, Moslehi J, Larkin J. Immune checkpoint inhibitors and cardiovascular toxicity. Lancet Oncol. 2018;19(9):e447–58. https://doi.org/10.1016/S1470-2045(18)30457-1.
Thompson JA, Schneider BJ, Brahmer J, Andrews S, Armand P, Bhatia S, et al. NCCN guidelines insights: management of immunotherapy-related toxicities, Version 1.2020. J Natl Compr Canc Netw. 2020;18(3):230–41. https://doi.org/10.6004/jnccn.2020.0012.
Velasco R, Villagran M, Jove M, Simo M, Vilarino N, Alemany M, et al. Encephalitis induced by immune checkpoint inhibitors: a systematic review. JAMA Neurol. 2021;78(7):864. https://doi.org/10.1001/jamaneurol.2021.0249.
Marini A, Bernardini A, Gigli GL, Valente M, Muniz-Castrillo S, Honnorat J, et al. Neurologic adverse events of immune checkpoint inhibitors: a systematic review. Neurology. 2021;96(16):754–66. https://doi.org/10.1212/WNL.0000000000011795.
Li Y, Zhang X, Zhao C. Guillain-Barré syndrome-like polyneuropathy associated with immune checkpoint inhibitors: a systematic review of 33 cases. BioMed Res Int. 2021;2021:1–17. https://doi.org/10.1155/2021/9800488.
Sechi E, Zekeridou A. Neurologic complications of immune checkpoint inhibitors in thoracic malignancies. J Thorac Oncol. 2021;16(3):381–94. https://doi.org/10.1016/j.jtho.2020.11.005.
Vogrig A, Muniz-Castrillo S, Joubert B, Picard G, Rogemond V, Skowron F, et al. Cranial nerve disorders associated with immune checkpoint inhibitors. Neurology. 2021;96(6):e866–75. https://doi.org/10.1212/WNL.0000000000011340.
Spain L, Walls G, Julve M, O’Meara K, Schmid T, Kalaitzaki E, et al. Neurotoxicity from immune-checkpoint inhibition in the treatment of melanoma: a single centre experience and review of the literature. Ann Oncol. 2017;28(2):377–85. https://doi.org/10.1093/annonc/mdw558.
Dalakas MC. Neurological complications of immune checkpoint inhibitors: what happens when you ‘take the brakes off’ the immune system. Ther Adv Neurol Disord. 2018;11:175628641879986. https://doi.org/10.1177/1756286418799864.
Thouvenin L, Olivier T, Banna G, Addeo A, Friedlaender A. Immune checkpoint inhibitor-induced aseptic meningitis and encephalitis: a case-series and narrative review. Ther Adv Drug Saf. 2021;12:204209862110047. https://doi.org/10.1177/20420986211004745.
Müller-Jensen L, Zierold S, Versluis JM, Boehmerle W, Huehnchen P, Endres M, et al. Characteristics of immune checkpoint inhibitor-induced encephalitis and comparison with HSV-1 and anti-LGI1 encephalitis: a retrospective multicentre cohort study. Eur J Cancer. 2022;175:224–35. https://doi.org/10.1016/j.ejca.2022.08.009.
Nannini S, Koshenkova L, Baloglu S, Chaussemy D, Noël G, Schott R. Immune-related aseptic meningitis and strategies to manage immune checkpoint inhibitor therapy: a systematic review. J Neurooncol. 2022;157(3):533–50. https://doi.org/10.1007/s11060-022-03997-7.
Daxini A, Cronin K, Sreih AG. Vasculitis associated with immune checkpoint inhibitors—a systematic review. Clin Rheumatol. 2018;37(9):2579–84. https://doi.org/10.1007/s10067-018-4177-0.
Kim JE, Patel K, Jackson CM. The potential for immune checkpoint modulators in cerebrovascular injury and inflammation. Expert Opin Ther Targets. 2021;25(2):101–13. https://doi.org/10.1080/14728222.2021.1869213.
Sundarrajan C, Bhai S, Dimachkie MM. Immune checkpoint inhibitor-related myositis: from pathophysiology to treatment. Clin Exp Rheumatol. 2022. https://doi.org/10.55563/clinexprheumatol/q7mdjs.
Fortes BH, Liou H, Dalvin LA. Ophthalmic adverse effects of immune checkpoint inhibitors: the Mayo Clinic experience. Br J Ophthalmol. 2021;105(9):1263–71. https://doi.org/10.1136/bjophthalmol-2020-316970.
Gan L, Chen H, Liu X, Zhang L. Ophthalmic immune-related adverse events associated with immune checkpoint inhibitors. Front Immunol. 2023;14:1130238. https://doi.org/10.3389/fimmu.2023.1130238.
Dinoto A, Mantovani E, Ferrari S, Mariotto S, Tamburin S. Cerebellar involvement associated with immune checkpoint inhibitors: a systematic review. Eur J Neurol. 2023;30(3):774–81. https://doi.org/10.1111/ene.15624.
Xu M, Nie Y, Yang Y, Lu Y-T, Su Q. Risk of neurological toxicities following the use of different immune checkpoint inhibitor regimens in solid tumors: a systematic review and meta-analysis. Neurologist. 2019;24(3):75–83. https://doi.org/10.1097/NRL.0000000000000230.
Michot JM, Lazarovici J, Tieu A, Champiat S, Voisin AL, Ebbo M, et al. Haematological immune-related adverse events with immune checkpoint inhibitors, how to manage? Eur J Cancer. 2019;122:72–90. https://doi.org/10.1016/j.ejca.2019.07.014.
Delanoy N, Michot J-M, Comont T, Kramkimel N, Lazarovici J, Dupont R, et al. Haematological immune-related adverse events induced by anti-PD-1 or anti-PD-L1 immunotherapy: a descriptive observational study. Lancet Haematol. 2019;6(1):e48–57. https://doi.org/10.1016/S2352-3026(18)30175-3.
Kramer R, Zaremba A, Mereira A, Ugurel S, Johnson DB, Hassel JC, et al. Hematological immune related adverse events after treatment with immune checkpoint inhibitors. Eur J Cancer. 2021;147:170–81. https://doi.org/10.1016/j.ejca.2021.01.013.
Ohashi T, Takase-Minegishi K, Meda A, Hamada N, Yoshimi R, Kirino Y, et al. Incidence and risk of hematological adverse events associated with immune checkpoint inhibitors: a systematic literature review and meta-analysis. J Hematol. 2023;12(2):66–74. https://doi.org/10.14740/jh1090.
Petrelli F, Ardito R, Borgonovo K, Lonati V, Cabiddu M, Ghilardi M, et al. Haematological toxicities with immunotherapy in patients with cancer: a systematic review and meta-analysis. Eur J Cancer. 2018;103:7–16. https://doi.org/10.1016/j.ejca.2018.07.129.
Zaremba A, Kramer R, De Temple V, Bertram S, Salzmann M, Gesierich A, et al. Grade 4 neutropenia secondary to immune checkpoint inhibition—a descriptive observational retrospective multicenter analysis. Front Oncol. 2021;11: 765608. https://doi.org/10.3389/fonc.2021.765608.
Mullally WJ, Cooke FJ, Crosbie IM, Kumar S, Abernethy VE, Jordan EJ, et al. Case report: thrombotic-thrombocytopenic purpura following ipilimumab and nivolumab combination immunotherapy for metastatic melanoma. Front Immunol. 2022;13: 871217. https://doi.org/10.3389/fimmu.2022.871217.
Neupane N, Shikhrakar S, Thapa S, Shrestha A. Hemophagocytic lymphohistiocytosis in patients treated with immune checkpoint inhibitors: a pharmacovigilance study from FDA Database. J Clin Oncol. 2023;41(16_suppl): e14684. https://doi.org/10.1200/JCO.2023.41.16_suppl.e14684.
Delyon J, Mateus C, Lambert T. Hemophilia A induced by ipilimumab. N Engl J Med. 2011;365(18):1747–8. https://doi.org/10.1056/NEJMc1110923.
Martin M, Nguyen H-Y, Beuvon C, Bene J, Palassin P, Atzenhoffer M, et al. Immune checkpoint inhibitor-related cytopenias: about 68 cases from the French pharmacovigilance database. Cancers. 2022;14(20):5030. https://doi.org/10.3390/cancers14205030.
Noseda R, Bertoli R, Müller L, Ceschi A. Haemophagocytic lymphohistiocytosis in patients treated with immune checkpoint inhibitors: analysis of WHO global database of individual case safety reports. J Immunother Cancer. 2019;7(1):117. https://doi.org/10.1186/s40425-019-0598-9.
Davis EJ, Salem J-E, Young A, Green JR, Brent Ferrell P, Ancell KK, et al. Hematologic complications of immune checkpoint inhibitors. Oncologist. 2019;24(5):584–8. https://doi.org/10.1634/theoncologist.2018-0574.
Gordon IO, Wade T, Chin K, Dickstein J, Gajewski TF. Immune-mediated red cell aplasia after anti-CTLA-4 immunotherapy for metastatic melanoma. Cancer Immunol Immunother. 2009;58(8):1351–3. https://doi.org/10.1007/s00262-008-0627-x.
Bennett R, Ruskova A. Atezolizumab-induced pure red cell aplasia. Br J Haematol. 2021;193(1):10–10. https://doi.org/10.1111/bjh.17259.
Guo Q, Zhao JN, Liu T, Gao J, Guo H, Cheng JM. Immune checkpoint inhibitor-induced aplastic anaemia: case series and large-scale pharmacovigilance analysis. Front Pharmacol. 2023;14:1057134. https://doi.org/10.3389/fphar.2023.1057134.
Helgadottir H, Kis L, Ljungman P, Larkin J, Kefford R, Ascierto PA, et al. Lethal aplastic anemia caused by dual immune checkpoint blockade in metastatic melanoma. Ann Oncol. 2017;28(7):1672–3. https://doi.org/10.1093/annonc/mdx177.
Schwab KS, Heine A, Weimann T, Kristiansen G, Brossart P. Development of hemolytic anemia in a nivolumab-treated patient with refractory metastatic squamous cell skin cancer and chronic lymphatic leukemia. Case Rep Oncol. 2016;9(2):373–8. https://doi.org/10.1159/000447508.
Leaf RK, Ferreri C, Rangachari D, Mier J, Witteles W, Ansstas G, et al. Clinical and laboratory features of autoimmune hemolytic anemia associated with immune checkpoint inhibitors. Am J Hematol. 2019;94(5):563–74. https://doi.org/10.1002/ajh.25448.
Smithy JW, Pianko MJ, Maher C, Postow MA, Shoushtari AN, Momtaz P, et al. Checkpoint blockade in melanoma patients with underlying chronic lymphocytic leukemia. J Immunother. 2021;44(1):9–15. https://doi.org/10.1097/CJI.0000000000000345.
Algaze SD, Park W, Harrington TJ, Mudad R. Autoimmune haemolytic anaemia in a patient with advanced lung adenocarcinoma and chronic lymphocytic leukaemia receiving nivolumab and intravenous immunoglobulin. BMJ Case Rep. 2018. https://doi.org/10.1136/bcr-2017-221801.
Wilson NR, Lockhart JR, Garcia-Perdomo HA, Oo TH, Rojas-Hernandez CM. Management and outcomes of hematological immune-related adverse events: systematic review and meta-analysis. J Immunother. 2022;45(1):13–24. https://doi.org/10.1097/CJI.0000000000000390.
Diaz L, Jauzelon B, Dillies A-C, Le Souder C, Faillie J-L, Maria ATJ, et al. Hemophagocytic lymphohistiocytosis associated with immunological checkpoint inhibitors: a pharmacovigilance study. J Clin Med. 2023;12(5):1985. https://doi.org/10.3390/jcm12051985.
Dupré A, Michot J-M, Schoeffler A, Frumholtz L, Baroudjian B, Delyon J, et al. Haemophagocytic lymphohistiocytosis associated with immune checkpoint inhibitors: a descriptive case study and literature review. Br J Haematol. 2020;189(5):985–92. https://doi.org/10.1111/bjh.16630.
La Rosée P, Horne A, Hines M, von Bahr Greenwood T, Manchowicz R, Berliner N, et al. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood. 2019;133(23):2465–77. https://doi.org/10.1182/blood.2018894618.
Liu LL, Skribek M, Harmenberg U, Gerling M. Systemic inflammatory syndromes as life-threatening side effects of immune checkpoint inhibitors: case report and systematic review of the literature. J Immunother Cancer. 2023;11(3): e005841. https://doi.org/10.1136/jitc-2022-005841.
Özdemir BC, Latifyan S, Perreau M, Fenwick C, Alberio L, Waeber G, et al. Cytokine-directed therapy with tocilizumab for immune checkpoint inhibitor-related hemophagocytic lymphohistiocytosis. Ann Oncol. 2020;31(12):1775–8. https://doi.org/10.1016/j.annonc.2020.08.2101.
Moore DC, Elmes JB, Arnall JR, Strassels SA, Patel JN. Immune checkpoint inhibitor-induced acquired haemophilia: a pharmacovigilance analysis of the FDA adverse event reporting system. Haemophilia. 2022. https://doi.org/10.1111/hae.14632.
Gidaro A, Palmieri G, Donadoni M, Mameli LA, La Cava L, Sanna G, et al. A diagnostic of acquired hemophilia following PD1/PDL1 inhibitors in advanced melanoma: the experience of two patients and a literature review. Diagnostics. 2022;12(10):2559. https://doi.org/10.3390/diagnostics12102559.
Fletcher J, Bird R, McLean AJW, O’Byrne K, Xu W. Acquired hemophilia a secondary to an immune checkpoint inhibitor: a case report. JTO Clin Res Rep. 2022;3(11): 100409. https://doi.org/10.1016/j.jtocrr.2022.100409.
Kato R, Hayashi H, Sano K, Handa K, Kumode T, Ueda H, et al. Nivolumab-induced hemophilia a presenting as gastric ulcer bleeding in a patient with NSCLC. J Thorac Oncol. 2018;13(12):e239–41. https://doi.org/10.1016/j.jtho.2018.06.024.
Gokozan HN, Friedman JD, Schmaier AH, Downes KA, Farah LA, Reeves HM. Acquired hemophilia A after nivolumab therapy in a patient with metastatic squamous cell carcinoma of the lung successfully managed with rituximab. Clin Lung Cancer. 2019;20(5):e560–3. https://doi.org/10.1016/j.cllc.2019.06.022.
Joly BS, Coppo P, Veyradier A. Thrombotic thrombocytopenic purpura. Blood. 2017;129(21):2836–46. https://doi.org/10.1182/blood-2016-10-709857.
De Filippis S, Moore C, Ezell K, Aggarwal K, Kelkar AH. Immune checkpoint inhibitor-associated thrombotic thrombocytopenic purpura in a patient with metastatic non-small-cell lung cancer. Cureus. 2021. https://doi.org/10.7759/cureus.16035.
Chiasakul T, Cuker A. Clinical and laboratory diagnosis of TTP: an integrated approach. Hematology. 2018;2018(1):530–8. https://doi.org/10.1182/asheducation-2018.1.530.
Lancelot M, Miller MJ, Roback J, Stowell SR. Refractory thrombotic thrombocytopenic purpura related to checkpoint inhibitor immunotherapy. Transfusion (Paris). 2021;61(1):322–8. https://doi.org/10.1111/trf.16117.
Nelson D, Kodsi M, Cockrell D, Morgan J, Key N. Thrombotic thrombocytopenic purpura associated with pembrolizumab. J Oncol Pharm Pract. 2022;28(4):979–82. https://doi.org/10.1177/10781552211073883.
Kozak M, Rubenstein W, Okwan-Duodu D, Friedman K, Nassir Y, Perez-Alvarez I, et al. Durable remission of thrombotic thrombocytopenic purpura in the setting of pembrolizumab therapy. Transfusion (Paris). 2023;63(6):1241–5. https://doi.org/10.1111/trf.17378.
Charvet E, Lheure C, Isnard C, Franck N, Kramkimel N, Vallet-Pichard A, et al. Hepatic sinusoidal obstruction syndrome induced by nivolumab in advanced melanoma: a case report. Ann Oncol. 2020;31(5):661–2. https://doi.org/10.1016/j.annonc.2020.02.004.
Ho TP, Venkatesh SK, Alkhatib HS, Yan Y. SOS! immunotherapy-associated sinusoidal obstructive syndrome. J Oncol Pract. 2019;15(12):675–6. https://doi.org/10.1200/JOP.19.00276.
Velev M, Baroudjian B, Pruvosit R, De Martin E, Laparra A, Babai S, et al. Immune-related generalised oedema—a new category of adverse events with immune checkpoint inhibitors. Eur J Cancer. 2023;179:28–47. https://doi.org/10.1016/j.ejca.2022.11.001.
Pi B, Wang J, Tong Y, Yang Q, Lv F, Yu Y. Immune-related cholangitis induced by immune checkpoint inhibitors: a systematic review of clinical features and management. Eur J Gastroenterol Hepatol. 2021;33(1S):e858–67. https://doi.org/10.1097/MEG.0000000000002280.
Albarrán-Artahona V, Laguna J-C, Gorría T, Torres-Jiménez J, Pascal M, Mezquita L. Immune-related uncommon adverse events in patients with cancer treated with immunotherapy. Diagnostics. 2022;12(9):2091. https://doi.org/10.3390/diagnostics12092091.
Zen Y, Yeh MM. Checkpoint inhibitor-induced liver injury: a novel form of liver disease emerging in the era of cancer immunotherapy. Semin Diagn Pathol. 2019;36(6):434–40. https://doi.org/10.1053/j.semdp.2019.07.009.
Patil PA, Zhang X. Pathologic manifestations of gastrointestinal and hepatobiliary injury in immune checkpoint inhibitor therapy. Arch Pathol Lab Med. 2021;145(5):571–82. https://doi.org/10.5858/arpa.2020-0070-RA.
Shalit A, Sarantis P, Koustas E, Trifylli E-M, Matthaios D, Karamouzis MV. Predictive biomarkers for immune-related endocrinopathies following immune checkpoint inhibitors treatment. Cancers. 2023;15(2):375. https://doi.org/10.3390/cancers15020375.
Mytareli C, Ziogas DC, Karampela A, Papalexis P, Siampanopoulou V, Lafioniatis A, et al. The uncharted landscape of rare endocrine immune-related adverse events. Cancers. 2023;15(7):2016. https://doi.org/10.3390/cancers15072016.
Tan MH, Iyengar R, Mizokami-Stout K, Yentz S, MacEachern MP, Shen LY, et al. Spectrum of immune checkpoint inhibitors-induced endocrinopathies in cancer patients: a scoping review of case reports. Clin Diabetes Endocrinol. 2019;5(1):1. https://doi.org/10.1186/s40842-018-0073-4.
He X, Tu R, Zeng S, He Z, Liu S, Fang Y. Non-bacterial cystitis secondary to pembrolizumab: a case report and review of the literature. Curr Probl Cancer. 2022;46(4): 100863. https://doi.org/10.1016/j.currproblcancer.2022.100863.
Li J, Yu Y-F, Qi X-W, Du Y, Li C-Q. Immune-related ureteritis and cystitis induced by immune checkpoint inhibitors: case report and literature review. Front Immunol. 2023;13:1051577. https://doi.org/10.3389/fimmu.2022.1051577.
Side Effect Registry Immuno-Oncology. 2023. [Online]. https://serio-registry.org/.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions.
Conflict of interest
LS has received advisory honoraria and speakers fees from Bristol Myers Squibb and Ipsen. Authors AJ, CB or AR declare that they have no conflicts of interest that might be relevant to the contents of this manuscript.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
No datasets were generated or analysed during the current study.
Code availability
Not applicable.
Authors contributions
AJ completed literature review, manuscript preparation, manuscript revision and prepared figures. CB completed literature review, manuscript preparation, manuscript revision and assisted with figure development. AR assisted with manuscript revision and figure development. LS assisted with overall design, manuscript revision, figure development and provided oversight of the project.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Javaid, A., Bennett, C., Rao, A. et al. Rare Immune-Related Adverse Events (irAEs): Approach to Diagnosis and Management. Pharm Med 38, 25–38 (2024). https://doi.org/10.1007/s40290-023-00508-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40290-023-00508-5