
Abstract
Background
ASC42 is a non-steroidal farnesoid X receptor agonist currently in clinical development for chronic liver diseases, such as nonalcoholic fatty liver disease/nonalcoholic steatohepatitis (NAFLD/NASH) and primary biliary cirrhosis (PBC).
Objective
The objective of this study was to assess the safety, pharmacokinetics (PK), and pharmacodynamics (PD) of ASC42 in healthy subjects.
Methods
We conducted the first-in-human study of ASC42 following single and multiple ascending doses (SAD/MAD) and food effect in healthy subjects. The SAD study included five cohorts receiving 5–200 mg ASC42 or placebo and one cohort that was given 15 mg ASC42 with a high-fat meal. The MAD study included three cohorts receiving 5–50 mg ASC42 or placebo once-daily (QD) for 14 days.
Results
A total of 65 healthy subjects were enrolled and one subject in the MAD study (cohort 8, ASC42 50 mg) withdrew from the study due to an unrelated serious adverse event (SAE) of atrial fibrillation. Pruritus was observed at the highest doses (200 mg cohort in SAD and 50 mg cohort in MAD). Most AEs were mild or moderate. No life-threatening or fatal AEs occurred. ASC42 showed a proportional increase in exposure and elimination half-life following both single and multiple dosing. There was a 21% and 37% decrease in area under the curve (AUC) and maximum plasma concentration (Cmax) when ASC42 was coadministered with food. The steady state was reached on day 4 with a mild accumulation (1.02–1.74-fold). ASC42 showed dose-dependent increases in fibroblast growth factor 19 and decreases in 7α-hydroxy-4-cholesten-3-one. Cholesterol remained within normal limits during study.
Conclusion
ASC42 was well tolerated with a pharmacokinetic profile suitable for QD dosing, and demonstrated dose-dependent targets engagement without altering plasma cholesterol in healthy subjects.
Trial registration number
NCT04679129.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
ASC42 had a favorable safety, and pharmacokinetic/pharmacodynamic profiles for further investigation. No pruritus was reported at therapeutic dose range (5–15 mg) of ASC42 in healthy subjects. |
1 Introduction
Farnesoid X receptor (FXR) agonists are under clinical investigation as potential targets for chronic liver diseases, including nonalcoholic steatohepatitis (NASH) and primary biliary cholangitis (PBC) [1]. Nonalcoholic fatty liver disease (NAFLD) is a metabolic stress liver injury closely related to insulin resistance and genetic susceptibility. NASH is regarded as the advanced form of NAFLD and responsible for progression to fibrosis, cirrhosis, and hepatocellular carcinoma. NASH is a condition characterized by the accumulation of fat in the liver (steatosis), inflammation, hepatocyte ballooning, and fibrosis [2]. PBC is a chronic, cholestatic liver disease characterized by the accumulation of bile acids in the liver, leading to cholestasis and subsequent progression to biliary fibrosis and cirrhosis [3]. Although NAFLD is the most common liver disease, with a worldwide prevalence of 25% [4], there are so far no approved therapies for NAFLD/NASH. Ursodeoxycholic acid (UDCA) is the recommended standard-of-care treatment for PBC, and has demonstrated enhanced liver transplant-free survival [5,6,7]; nevertheless, 30–40% of all UDCA-treated patients lack satisfactory responses to UDCA, which substantially increases the risk of death or need for liver transplantation [7].
FXR is a key nuclear receptor predominantly expressed in the liver and gut, which controls intricate signaling pathways associated with the regulation of bile acid homeostasis. FXR has been identified as the bile acid sensor. FXR activation triggers a delicate negative feedback mechanism that regulates several facets of bile acid metabolism directly in target organs or indirectly via fibroblast growth factor 19 (FGF19) [8], a naturally occurring hormone released from enterocytes in response to physiological FXR agonism, resulting in reduced bile acid levels. In addition, FXR activation modulates expression of genes involved in bile acid transport and metabolism as well as glucose and lipid metabolism. Thus, the therapeutic use of FXR agonists to modify hepatic metabolism is anticipated to bring benefits to patients with NAFLD/NASH or PBC. In fact, obeticholic acid (OCA), a bile acid-derived FXR agonist, has been approved in combination with UDCA for PBC patients with an inadequate response to UDCA or as monotherapy in patients intolerant of UDCA [9]. Furthermore, recent studies have expanded OCA use for NASH treatment [10]. Phase 2 studies (FLINT) have demonstrated improvements in histology [11], while recent data from the phase 3 study (REGENERATE) showed that OCA achieved statistically significant improvement in at least 1 stage of fibrosis without worsening of NASH [12].
The FLINT study found that OCA led to improvements in important histological characteristics associated with NASH, such as fibrosis. However, complete resolution of NASH was not observed. Moreover, 23% of patients reported pruritus, and OCA-treated patients experienced significant increases in total and low-density lipoprotein cholesterol (TC and LDL-C), along with a minor decrease in high-density lipoprotein cholesterol (HDL-C).
Nevertheless, pruritus, constipation, diarrhea, and hyperlipidemia were significant events with a clear dose-dependent effect that are often reported with OCA, and it can have a negative impact on tolerability. Thus, there is still a need to develop more effective FXR agonists with improved pharmacokinetics (PK) and safety profiles to successfully manage NAFLD/NASH and PBC.
ASC42 is a novel non-steroidal selective FXR agonist. Based on promising preclinical validation results of ASC42 in animal models of NASH and favorable preclinical safety assessments, this first-in-human (FIH) study was conducted to evaluate safety and tolerability pharmacokinetics (PK) and pharmacodynamics (PD) of ASC42 following single ascending doses under fasting or fed conditions and multiple ascending doses in healthy subjects.
2 Methods
This study was conducted in ICON Research Center and approved by the institute research board committee. All procedures performed in studies were in accordance with the Declaration of Helsinki and International Conference on Harmonisation Good Clinical Practice. Written informed consent was obtained from each subject at the screening prior to the initiation of any study-related procedures.
This randomized first-in-human double-blind placebo-controlled phase I study consisted of two parts: (1) single ascending doses (SAD) in five cohorts (part Ia) with an additional food effect cohort (part Ib) and (2) multiple ascending doses (MAD) in three cohorts (part II) in healthy subjects.
2.1 Study Population
Major inclusion criteria for this study were healthy subjects aged 18–65 years, weighed at least 50 kg, provided written informed consent, and had physical examination and vital signs within normal range or slightly abnormal but, in the opinion of the investigator, had no clinical significance. Key exclusion criteria included women of childbearing potential; any surgical or medical condition that could significantly alter the absorption, metabolism, distribution, and excretion of the study drug; medical history and/or clinical or laboratory evidence of liver disease or liver injury as indicated by abnormal liver tests, such as alanine aminotransferase (ALT), aspartate aminotransferase (AST), γ‐glutamyl transferase (GGT), alkaline phosphatase, or serum bilirubin levels exceeding 1× the upper limit of normal (ULN); or a positive hepatitis B surface antigen or hepatitis C test result.
2.2 Study Design
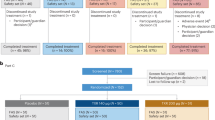
Study design is shown in Fig. 1. In part Ia SAD study, eight subjects per cohort were randomized to receive a single dose of ASC42 (5, 15, 50, 100, or 200 mg) or placebo in a 3:1 ratio. In part Ib food effect study, cohort 2 (15 mg) was selected to evaluate the food effect on the PK of ASC42 since the dose level is close to the predicted human efficacious dose (10 mg). In part II MAD study, eight subjects per cohort were randomized to receive ascending once daily (QD) doses of ASC42 (5, 15, or 50 mg) or placebo in a 3:1 ratio for 14 days.

Study design. This study consisted of two parts: SAD in five cohorts (part Ia) with an additional food effect cohort (part Ib) and MAD in three cohorts (part II). Each cohort consisted of eight subjects (six active, two placebo)
2.3 Randomization
A randomization list was produced using a validated system that automated the random assignment of treatment arms to randomization numbers in the specified ratio.
2.4 PK Assessments
Plasma concentrations of ASC42 were measured by a validated liquid chromatography tandem mass spectrometry (LC–MS/MS) method. Calibration curves were obtained using a 1/concentration2 weighted linear regression. Standards and quality control samples were within 20% of nominal values with a dynamic range of 0.200–100 ng/mL. Assay accuracy and precision of standards and quality controls were within 5% and 10%, respectively. Blood samples for PK assessment were collected at different timepoints as protocol specified. Samples of high concentration (> 100 ng/mL) were diluted for quantitative analysis. Based on concentration–time profiles of ASC42, the following PK parameters would be calculated: (1) SAD study—maximum plasma concentration (Cmax) and time to reach maximum plasma concentration following drug administration (Tmax), area under the concentration–time curve from time 0 to 24-h post dose (AUC0–24h), AUC from time 0 to the time of the last quantifiable concentration (AUClast), AUC from time 0 to infinity (AUCinf), percentage of AUCinf extrapolated (%AUCext), apparent total body clearance after oral administration (CL/F), apparent volume of distribution after oral administration (Vd/F), and terminal elimination half-life (T1/2) under the fasted condition; (2) MAD study—Cmax, minimum observed concentration (Cmin), trough concentration (Ctrough), Tmax, time to reach minimum plasma concentration following drug administration (Tmin), AUC over one dosing interval for multiple dose (AUCτ), %AUCext, CL/F at steady state (CL/Fss), Vd/F at steady state (Vd/Fss), the effective elimination half-life (T1/2,eff), accumulation ratio for Cmax (RCmax), and accumulation ratio for AUCτ (RAUC) under the fasted condition; and (3) Cmax, Tmax, AUClast, AUCinf, CL/F, Vd/F, and T1/2 under the fed condition.
2.5 PD Assessments
PD assessments were evaluated by the changes of PD biomarkers, FGF19 and 7α‐hydroxy‐4‐cholesten‐3‐one (C4), from the baseline. Serum FGF19 was measured by enzyme-linked immunosorbent assay (ELISA) (FGF19 Quantikine Enzyme-Linked Immunosorbent Assay Kit, R&D Systems, Minneapolis, MN). The validated concentration range was 15.6–1000 pg/mL. Serum C4 was measured using LC–MS/MS. The validated concentration range was 0.5–200 ng/mL. Blood samples for PD assessment were collected at different timepoints as the protocol specified. Maximum effect (Emax), maximum change from baseline effect (ECFB), and area under the effect curve from time 0–24 h (AUEC0–24h) for FGF19 and minimum effect (Emin), ECFB, and AUEC0–24h for C4 would be calculated. Sigmoid Emax model was used in PD assessment.
2.6 Safety and Tolerability Assessments
Safety was monitored throughout the study using standard measures including: adverse event (AE) monitoring, clinical laboratory testing, vital signs, physical examinations, and 12-lead electrocardiograms (ECGs) and continuous ECG recordings. The safety analysis set included all subjects who received at least one dose of the study drug or matching placebo.
2.7 Sample Size and Statistics
The sample size of eight subjects per cohort in both part I (SAD) and part II (MAD) is typical in first-in-human studies and was deemed sufficient to meet the objective of safety tolerability and PK/PD assessment of each cohort. The sample size was not based on statistical power considerations.
Data analyses were performed using SAS 9.4. PK parameters were calculated through non-compartmental analysis (NCA) using Phoenix™ WinNonlin™ 8.0 (Certara, Princeton, NJ). Sigmoid Emax model was used in PD assessment. Comparisons between fasted and fed conditions (food effect) were evaluated by a mixed-effect analysis of variance (ANOVA) of log-transformed ASC42 PK parameters (AUClast, AUCinf, and Cmax). Absence of food effect was to be concluded if the least square (LS) GM ratio of ASC42 PK parameters (AUClast, AUCinf, and Cmax) and the corresponding 90% confidence interval (CI) fell within limits 80–125%. LS linear regression model was used to determine whether ASC42 PK parameter was dose proportional. AEs would be classified by system organ class (SOC) and preferred terms (PT) according to the MedDRA Version 24.0. Increase of FGF19 and reduction of C4 on day 1 for SAD study and on days 1 and 14 for MAD study compared with baseline (pre-dose on day 1) at different dose levels would be calculated. The Wilcoxon rank sum test was performed to compare the median difference of changes from baseline in FGF19 and C4 concentrations between each ASC42 dose and the pooled placebo group. A two-sided p value less than 0.05 was considered statistically significant.
For PK analysis, concentrations below the lower limit of quantitation (LLOQ) were considered as 0 in summary statistics. While for PK parameter calculations, concentrations below the LLOQ were considered as 0 (before Tmax) or missing (after Tmax). For PD biomarkers, values below LLOQ were replaced by LLOQ/2 in analyses and for PD parameter calculations.
3 Results
3.1 Subject Demographics and Disposition
In total, 65 healthy subjects were enrolled. All subjects in both part I (SAD) and part II (MAD) received their planned dose except for one subject in part II (cohort 8, ASC42 50 mg) who discontinued the study drug following 3 out of 14 planned daily doses (treatment compliance of 21.4%) due to an unrelated serious adverse event (SAE) of atrial fibrillation and who was replaced by another eligible subject; the eligible subject was followed until day 35. No significant protocol deviations were recorded in this study. All 65 subjects were included for safety evaluation, while the 64 subjects who completed all dosing and follow-up visits were included for pharmacokinetics and pharmacodynamics evaluation.
Demographics and baseline characteristics are shown in Table 1. In the MAD study, more subjects were male. In the SAD study, median age ranged from 32 to 56.5 years and 31–48.5 years in ASC42 and placebo groups, respectively. In the MAD study, age ranged from 24 to 62 years in ASC42 groups and 32–54 years in placebo groups. All treatment groups in the SAD and MAD studies were comparable regarding body mass index (BMI), except for the placebo group in food effect study cohort 2, who had a slightly lower median BMI of 22.1 kg/m2.
3.2 Safety and Tolerability Assessments
Treatment-emergent adverse events (TEAEs) by SOC and PT are summarized in Table 2. Overall, seven subjects (17.5%) in part I (SAD) reported eight TEAEs, one fed subject (12.5%) in cohort 2 reported one TEAE, and nine subjects (36.0%) in part II (MAD) reported thirteen TEAEs. Most AEs were mild (grade 1) or moderate (grade 2) in severity. Only two subjects (8.0%) in part II (MAD) reported two severe (grade 3) AEs of pruritus, which all occurred at 50 mg dosing group (cohort 8). No life-threatening or fatal AE was reported. Five subjects (12.5%) in part I (SAD), one fed subject (12.5%) in cohort 2, and five subjects (20.0%) in part II (MAD) had study drug-related AEs. Notably, four of the five subjects in part II with study drug-related AEs were in cohort 8 (ASC42 50 mg) and accounted for seven of the eight total study drug-related TEAEs in part II. One subject receiving placebo (10.0%) in part I (SAD) and one subject receiving placebo (16.7%) in part II (MAD) reported one AE each and both AEs were considered as placebo-related. The most frequently reported AE in part I (SAD) was headache, occurring as one event each in three subjects overall (7.5%). The most frequently reported AE in part II (MAD) was pruritus, reported as one event each in four subjects (57.1%), and all were in cohort 8. One SAE of atrial fibrillation was reported in part II. This subject is a 47-year-old Black or African American male who had normal ECG results at screening, with the exception of the interpretation of sinus bradycardia with left ventricular hypertrophy and ST–T change (voltage criteria plus ST–T abnormality) that was considered abnormal, but not clinically significant, by the Investigator. The ECG conducted pre-dose on day 4 indicated the clinically significant abnormality of atrial fibrillation, then normal sinus rhythm was achieved after one synchronized defibrillation after being relocated to a hospital emergency room. Investigator and consulting cardiologist revealed potential lifestyle and/or life events may have contributed to a prestudy medical history of paroxysmal atrial fibrillation, undiagnosed prior to enrollment in this study. Together with subject’s retrospective reported medical history of intermittent chest tightness and history of benign ethnic neutropenia, the atrial fibrillation was subsequently presumed to be related to an undiagnosed preexisting condition and therefore unlikely related to study drug. This SAE led to the subject withdrawal on day 4 (three doses received) and the patient was followed until day 35. There were sporadic clinically significant abnormalities in clinical laboratory results, vital signs, physical examinations, and ECGs but they were not clinically meaningful and no trends were identified.
No AE of pruritus was reported in cohorts below 200 mg dose level in SAD study and in cohorts below 50 mg dose level in MAD study, thus ASC42 was safe and well tolerated when administered as single doses up to 100 mg and multiple daily doses for 14 days up to 15 mg in healthy subjects.
3.3 PK Assessments
Mean ± standard deviation (SD) ASC42 plasma concentration–time profiles are presented in Fig. 2. In part I (SAD) study, following a single dose of ASC42 under fasted condition, ASC42 concentration increased with increasing dose (5–200 mg doses, Fig. 2A). Mean concentrations peaked between 2 and 4 h and then declined biexponentially. Concentrations were detectable in all but one subject (5 mg dose) through 48 h post-dose and to 72 h post-dose for the majority of subjects in the 50, 100, and 200 mg dose cohorts. Mean plasma concentrations were slightly lower and peaked slightly later when ASC42 was administered with food compared with administration under fasting condition (Fig. 2B). In part II (MAD) study, following multiple doses of ASC42, ASC42 concentration increased with an increasing dose (5–50 mg doses, Fig. 2C). Mean concentrations peaked between 3 and 4 h. Concentrations were detectable in all subjects throughout the 24-h sampling period. Mean pre-dose concentrations (Ctrough) showed that ASC42 concentrations appeared to reach steady state by day 4 (Fig. 2C).

Pharmacokinetic profile of ASC42 following single and multiple ascending doses. A Mean plasma concentration–time profiles following single oral doses of ASC42 under fasting conditions (semilogarithmic scale). B Mean plasma concentration–time profiles following a single oral dose of 15 mg ASC42 under fed and fasted conditions (linear scale). C Mean plasma concentration–time profiles following multiple ascending doses of ASC42 for 14 days (linear scale). Data are represented as mean ± standard error
ASC42 PK parameters are summarized in Table 3. An assessment of the food effect on ASC42 PK and dose proportionality of ASC42 are shown in Supplementary Tables 1 and 2, respectively. Intersubject variability (GeoCV% variabilities) was observed for Cmax and AUCs ranging from 20% to 122% across the 5–200 mg doses, with the highest variabilities in the 5 mg and 200 mg dose groups. Both Cmax and AUCs increased with increasing dose of ASC42. The median Tmax ranged from 1.75 to 4 h (individual range of 1.5–10.0 h). The median Tmax was slightly later (5.0 h) when ASC42 was administered with food (individual range of 4.00–6.00 h). This suggested that food delayed the gastric emptying and intestinal transport time of the drug, thereby affecting Tmax of the drug absorption. The t1/2 was similar (approximately 8 h) for the 15–200 mg doses and slightly longer for the 5 mg dose (11 h). There was a 21% and 37% decrease in geometric least squares mean (LSM) AUC and Cmax, respectively, following a single 15 mg dose given with food compared with when given under fasting condition. This suggested that food increased blood flow to the liver, which accelerated drug metabolism. The 90% CI for geometric LSM ratio AUClast, AUCinf, and Cmax with fasting versus fed did not fall within the prespecified limits of 80–125%. The lower bound of the 95% CI for the slope of the regression line for each PK parameter was greater than 1. Therefore, it can be concluded that the PK of ASC42 was more-than-dose-proportional across the range of 5–200 mg following single dose administration under fasting conditions. This might be due to metabolic enzyme saturation with the dose increasing.
For MAD study, Cmax and AUCτ increased with increasing dose of ASC42 following a single dose on day 1 or QD doses for 14 days. Geometric CV% for Cmax and AUCτ ranged from 52 to 109%. The median Tmax was 3–4 h. The effective t1/2 following 14 days of QD dosing was similar for the 5 and 15 mg doses (11–13 h) and higher for the 50 mg dose (23 h). Geometric mean CL/Fss following 14 days of QD dosing decreased with increasing dose. The accumulation ratios based on day 14 to day 1 AUC0–τ and Cmax ranged from 1.00 to 1.74 and 1.00 to 2.23, respectively, for 5–50 mg ASC42 administered QD for 14 days. The ratio for the 15 mg ASC42 dose was approximately 1.3. The lower bound of the 95% CI for the slope of the regression line for each parameter was greater than 1 (Supplementary Table 3). Therefore, it can be concluded that the PK of ASC42 was overdose proportional across the range of 5–50 mg following multiple dose administration under fasting conditions. This suggested that metabolic enzyme saturation may have occurred.
3.4 PD Assessments
Major PD parameters of FGF-19 and C4 following multiple doses of ASC42 are summarized in Table 4. Drug‐associated elevations in mean FGF19 and declines in mean C4 were observed. For FGF19, the mean Emax, AUC0–24, AUEC0–24 Emax/baseline, and ECFB for subjects treated with ASC42 were higher than placebo in nearly all instances (except for 5 mg, similar FGF19 exposure was seen compared with the placebo) and appeared to increase with increasing dose. Baseline was similar for day 1 and day 14; therefore, parameters corrected for baseline were similar following either single or multiple doses. For C4, the mean Emin, AUC0–24, AUEC0–24, Emin/baseline, and ECFB for subjects treated with ASC42 were lower than placebo in nearly all instances and appeared to decrease with increasing dose. On day 14, the concentrations of C4 decreased more significantly as compared with day 1 in all dose groups. Overall, exposure to ASC42 increased with increasing dose and there was an increase in FGF19 and decrease in C4 parameters.
3.5 Effects of ASC42 on Cholesterol
TC, HDL-C, and LDL-C remained within normal limits during the study (Fig. 3). Mean HDL-C of 50 mg in MAD study was a little below the normal limit on day 14, but back to normal on day 21.

Effect of ASC42 on cholesterol. Total cholesterol (TC), high-density lipoprotein cholesterol (HDL-C), and low-density lipoprotein cholesterol (LDL-C) remained within normal limits during 14-day ASC42 treatment
4 Discussions
Results of this phase I study in healthy subjects showed that PK of ASC42 was overdose proportional across the range of 5–200 mg single dose administration under fasting conditions, or across the range of 5–50 mg multiple dose administration. Median Tmax ranged from 1.75 to 4.00 h following a single dose of ASC42 on day 1 and was slightly later following administration with food (5.00 h) and the t1/2 was similar (approximately 8 h) for the 15–200 mg doses and slightly longer for the 5 mg dose (11 h). There was a 21% and 37% decrease in AUC and Cmax, respectively, when ASC42 was taken with food compared with when given in the fasted state, indicating a minimal food effect. The accumulation ratio based on day 14 to day 1 AUC0–τ and Cmax ranged from 1.02 to 1.74 and 1.00 to 2.23, respectively, for 5–50 mg ASC42, indicating a mild accumulation. The accumulation ratio for the 15 mg ASC42 dose (proposed the highest therapeutic dose) was low, approximately 1.3.
Concentrations of FGF-19 in subjects receiving ASC42 were all higher than those receiving placebo (with the exception of the 5 mg single dose) and appeared to increase with increasing doses of ASC42. The concentrations of C4 were all lower in subjects receiving ASC42 than those receiving placebo and appeared to decrease with increasing doses of ASC42. C4 is a metabolic intermediate in the rate limiting step for the synthesis of bile acids from hepatic cholesterol. FGF19 is a hormone released by ileal enterocytes after stimulation of nuclear FXRs, typically by absorbed bile acids, and FGF19 provides negative feedback for bile acid synthesis in hepatocytes. Thus, increase of FGF19 and decrease of C4 indicated impediment of bile acid synthesis, suggesting the therapeutic potential of ASC42 for NAFLD and PBC.
Based on both mouse and rat NASH animal models, the predicted human therapeutic dose was 15 mg once daily (unpublished data). This phase I study showed that during 14-day treatment of 15 mg once daily day 1 and day 14, FGF19 levels were 13.5 and 17.80-fold of the pre-dose baseline, respectively; day 1 and day 14 C4 levels decreased 84% and 91% from the baseline, respectively. Based on these data, 15 mg once-daily dose could be considered as the highest therapeutic dose for future studies. The magnitude of FGF19 increase and/or C4 decrease may be used to project potential levels of liver fat reduction in NASH or PBC patients with a ≥ 30% relative liver fat reduction on magnetic resonance imaging-derived proton density fat fraction (MRI-PDFF) potentially correlating with an increased likelihood of histologic benefit [13, 14].
Results from other clinical trials in NASH and PBC have shown that OCA and other FXR agonists cause class-related side effects, including pruritus, increased plasma cholesterol levels, and reduced levels of HDL-C [15]. Only pruritus was reported in this study, and occurred only at the highest doses. Most AEs were mild (grade 1) or moderate (grade 2) in severity, and only two subjects (8.0%) in part II (MAD) reported two severe AEs of pruritus. No life-threatening or fatal TEAEs occurred. The most common AE in part II (MAD) was pruritus, reported as one event each in four subjects (57.1%) in cohort 8. Pruritus was also reported for one subject in cohort 5 (ASC42 200 mg) of part I. ASC42 did not cause pruritus in healthy subjects up to 3 weeks following daily dosing up to 15 mg for 2 weeks. Slight reduction of HDL-C was observed in the highest multiple doses of 50 mg ASC42 group on day 14. However, since there was no significant difference and the highest therapeutic dose would be selected as 15 mg or lower daily dosing, the potential HDL-C reduction would not be relevant. It was noticed that the exposure at 50 mg daily dosing was 6.2-fold higher than that of 15 mg daily dosing as the dose increased by 3.3 fold, indicating a reasonable safety margin (6.2) for pruritus. Although one SAE was observed in part II (cohort 8, ASC42 50 mg), it was considered unlikely related to the ASC42 due to potential lifestyle and/or life event, which may have contributed to a pre-study medical history of paroxysmal atrial fibrillation, undiagnosed prior to enrollment in this study.
There were some limitations in this study. Due to only having a 14-day treatment duration, the drug‐induced pruritus needs to be further evaluated with the longer treatment duration. Effect of ASC42 on TC, HDL-C, and LDL-C levels also need to be evaluated with the longer treatment duration.
5 Conclusions
ASC42 was in general safe and well tolerated when administered as single doses up to 100 mg and multiple daily doses for 14 days up to 15 mg in healthy subjects. ASC42 at therapeutic dose range (5–15 mg) had an acceptable safety profile and showed no drug‐induced pruritus or transient elevations in serum ALT, AST, or GGT. Furthermore, ASC42 showed effective FXR target engagement in dose‐dependent elevations in FGF19 and reduction in C4, and caused no significant changes in TC, HDL-C, and LDL-C levels in healthy subjects up to 3 weeks following daily dosing for 14 days. Results of this study support the continued investigation of ASC42 in patients with NAFLD/NASH/PBC. ASC42 is currently in a 12-week phase 2 trial to treat PBC.
References
Gege C, Hambruch E, Hambruch N, Kinzel O, Kremoser C. Nonsteroidal FXR ligands: current status and clinical applications. Handb Exp Pharmacol. 2019;256:167–205.
Parthasarathy G, Revelo X, Malhi H. Pathogenesis of nonalcoholic steatohepatitis: an overview. Hepatol Commun. 2020;4(4):478–92.
Gulamhusein AF, Hirschfield GM. Primary biliary cholangitis: pathogenesis and therapeutic opportunities. Nat Rev Gastroenterol hepatol. 2020;17(2):93–110.
Cotter TG, Rinella M. Nonalcoholic fatty liver disease 2020: the state of the disease. Gastroenterol. 2020;158(7):1851–64.
Poupon RE, Poupon R, Balkau B. UDCA-PBC Study Group Ursodiol for the long-term treatment of primary biliary cirrhosis. N Engl J Med. 1994;330:1342–7.
Poupon RE, Balkau B, Eschwège E, Poupon R. UDCA-PBC Study Group A multicenter, controlled trial of ursodiol for the treatment of primary biliary cirrhosis. N Engl J Med. 1991;324:1548–54.
Harms MH, van Buuren HR, Corpechot C, Thorburn D, Janssen HLA, Lindor KD, et al. Ursodeoxycholic acid therapy and liver transplant-free survival in patients with primary biliary cholangitis. J Hepatol. 2019;71:357–65.
Kliewer SA, Mangelsdorf DJ. Bile acids as hormones: the FXR-FGF15/19 pathway. Dig Dis (Basel, Switzerland). 2015;33(3):327–31.
Brown Jr RS. Use of obeticholic acid in patients with primary biliary cholangitis. Gastroenterol Hepatol. 2018;14(11):654–7.
Chew NW, Ng CH, Truong E, Noureddin M, Kowdley KV. Nonalcoholic steatohepatitis drug development pipeline: an update. Semin Liver Dis. 2022;42:379–400.
Neuschwander-Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. 2015;385:956–65.
Younossi ZM, Ratziu V, Loomba R, Rinella M, Anstee QM, Goodman Z, et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet. 2019;394:2184–96.
Zöhrer E, Alisi A, Jahnel J, et al. Efficacy of docosahexaenoic acid-choline-vitamin E in paediatric NASH: a randomized controlled clinical trial. Appl Physiol Nutr Metab. 2017;42(9):948–54.
Li Z, Lin B, Lin G, et al. Circulating FGF19 closely correlates with bile acid synthesis and cholestasis in patients with primary biliary cirrhosis. PLoS One. 2017;12(6): e0178580.
Panzitt K, Zollner G, Marschall HU, Wagner M. Recent advances on FXR-targeting therapeutics. Mol Cell Endocrinol. 2022;552: 111678.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was sponsored by Gannex Pharma Co., Ltd. Gannex Pharma was involved with the study design, statistical analysis, interpretation of data, decision to publish, and preparation of the manuscript.
Conflict of Interest
H.H. and J.J.W. are employees and stock/shareholders of Gannex Pharma.
Ethics approval
This study was conducted in conformance with good clinical practice (GCP) standards and applicable country and/or local statutes and regulations regarding ethical committee review, informed consent, and the protection of human subjects participating in biomedical research.
Consent to participate
Written informed consent was obtained from each subject at the screening visit prior to the initiation of any study-related procedures. The investigator or designee explained the study procedures, risks, and potential benefits if any. Subjects reviewed the study instructions and informed consent form (ICF), and were given the time and opportunity to have any questions concerning the conduct of the study, with all answered to their satisfaction.
Consent for publication
Not applicable.
Availability of data and material
Any additional information required to reanalyze the data reported in this paper is available from the corresponding author upon request from the publication of the paper. Requests for data sharing will be responded to within 2–3 weeks.
Authors’ contributions
Jinzi J. Wu: design and conduct of the study including safety surveillance; interpretation of results; critical review and revision of the manuscript; and approval of the final manuscript for submission. Handan He: PK, PD, and biomarker statistical analyses; interpretation of results; and critical review and revision of the manuscript.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
He, H., Wu, J.J. Human Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of ASC42, a Novel Farnesoid X Receptor Agonist. Drugs R D 23, 453–464 (2023). https://doi.org/10.1007/s40268-023-00444-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-023-00444-4