
Abstract
Background
The antibacterial agent prulifloxacin, a prodrug of ulifloxacin, is indicated in the treatment of acute lower urinary tract infections, acute exacerbation of chronic bronchitis and acute bacterial rhinosinusitis.
Objective
We aimed to provide new insights on the pharmacokinetics (PK) of ulifloxacin in patients with different degrees of renal impairment.
Methods
A two-site, international, open-label, parallel-group, single- and repeated-dose study was performed. The drug was administered as a single dose of 600 mg to subjects with normal renal function and patients with mild, moderate and severe renal impairment. Subsequently, the same dose was administered daily for 7 days to subjects with normal renal function and patients with mild and moderate renal impairment, while a dose of 300 mg was administered daily for 7 days to patients with severe renal impairment. Plasma and urine ulifloxacin levels were measured. Complete safety evaluation was performed.
Results
Exposure to ulifloxacin increased as renal function decreased due to a lower ulifloxacin clearance. Ulifloxacin PK were significantly changed only in patients with severe renal impairment. The amount of ulifloxacin excreted in urine over a 24-h dosing period was similar in subjects with normal renal function and patients with mild impaired renal function, but lower in those with moderate and severe renal impairment.
Conclusion
Our data show that prulifloxacin is a safe quinolone and is well tolerated in both subjects with normal renal function and patients with impaired renal function, requiring a minimal dosage adjustment only in patients with severe renal impairment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
This study clearly demonstrates that the mean exposure to ulifloxacin was comparable between renally impaired subjects and subjects with normal renal function, while maintaining the daily dose of 600 mg prulifloxacin for patients with mild and moderate renal impairment and reducing the daily dose to 300 mg for subjects with severe renal impairment. |
Prulifloxacin was safe and well tolerated upon both single and multiple dosing in all subject groups. |
1 Introduction
Prulifloxacin is the prodrug of ulifloxacin, an antibacterial agent of the fluoroquinolone group [1]. Due to its broad spectrum of activity against both Gram-negative and Gram-positive bacteria, it is specifically indicated for the treatment of acute uncomplicated lower urinary tract infections (UTIs), complicated lower UTIs, acute exacerbation of chronic bronchitis (AECB) and acute bacterial rhinosinusitis (ABRS) [2,3,4]. Indeed, after absorption from the gastrointestinal tract, prulifloxacin is immediately and almost quantitatively metabolized to the active metabolite ulifloxacin [5] before entering the systemic circulation, showing a good penetration into peripheral target tissues [6]; no other known potentially active metabolites are formed [7]. Approximately 45% of ulifloxacin is bound to serum proteins in vivo, reaching concentrations in target organs approximately fivefold higher than in plasma [8]. The elimination half-life of ulifloxacin after the administration of single doses of prulifloxacin 600 mg is approximately 9–12 h in healthy subjects [7]. Ulifloxacin undergoes negligible phase I metabolism, and is mainly eliminated unchanged, partially as glucuronate, through the urine and faeces via biliary excretion. Elimination occurs primarily through the faeces and accounts mainly for the unabsorbed fraction of the dose, and then to the amount excreted through the bile, which does not undergo reabsorption in the intestine. Excluding the contribution of the unabsorbed fraction of the dose, renal elimination appears equal to/higher than faecal elimination. Therefore, the clearance of unchanged ulifloxacin through direct renal excretion is an important way of elimination of prulifloxacin [7].
Currently, the recommended dosage regimens in adults are one single prulifloxacin 600 mg tablet in patients with acute uncomplicated UTIs, and up to a 10-day treatment period with prulifloxacin 600 mg once daily in complicated UTIs, AECB and ABRS [3, 4].
Prulifloxacin is also used to treat infections (i.e. UTIs, AECB and ABRS) that often occur in an elderly and fragile population with impaired renal function [9, 10]. However, the lack of pharmacokinetic (PK) data for prulifloxacin in patients with different degrees of renal impairment prevents the determination of a correct prulifloxacin dosage for this group of patients. Hence, the dosage adjustment in patients with renal impairment is currently complex and time-consuming as it requires thorough monitoring of ulifloxacin plasma levels after drug administration. To overcome these issues, a clinical trial targeting patients with renal impairment was designed to define the ulifloxacin PK at steady state. The PK data we obtained are the key to establishing the correct prulifloxacin dosage for the treatment of patients with different degrees of renal impairment.
2 Methods
2.1 Patients and Study Design
We performed a two-site, international, open-label, parallel-group, single (phase A) and repeated (phase B) dose study, where phases A and B are separated by a washout period of at least 1 week, during which an interim analysis was carried out. The study was specifically designed to assess the influence of renal impairment, compared with healthy subjects, on the following plasma PK parameters of ulifloxacin after single and repeated oral dose administration of prulifloxacin:
-
AUCt: area under the plasma concentration-time curve from time 0 to the time of last quantifiable concentration;
-
Cmax: maximum plasma concentration;
-
Cmax,ss: maximum plasma concentration at steady state;
-
t½: apparent terminal elimination half-life;
-
AUCτ,ss: area under the plasma concentration-time curve during a dosing interval at steady-state;
-
AUC∞: area under the plasma concentration-time curve extrapolated to infinity;
-
tmax,ss: time to maximum plasma concentration at steady state.
The study was undertaken in South Africa and Germany. The first subject was screened on 28 March 2012, and the last subject was completed on 25 October 2013. The study conformed to the ethical principles set out in the Declaration of Helsinki and was approved by the regulatory authorities and independent Ethics Committees in the countries participating in this trial. All patients provided written informed consent before study entry.
Eligible subjects were men or women aged 18–75 years suffering from renal impairment, whose extent was measured by the estimated glomerular filtration rate (eGFR). Subjects were divided into four groups, according to the eGFR value: eGFR > 80 mL/min: normal renal function; eGFR 50–80 mL/min: mild renal impairment; eGFR 30 to < 50 mL/min: moderate renal impairment; eGFR < 30 mL/min: severe renal impairment. The eGFR value was assessed with the use of the endogenous marker creatinine via the Cockroft–Gault formula, as commonly used in clinical practice, and confirmed by the sinistrin clearance method. The sinistrin GFR (sGFR) was used to establish a relationship between GFR and ulifloxacin clearance.
The major exclusion criteria were end-stage renal disease with current or past dialysis, moderate to severe liver function impairment, positive test results for hepatitis B/C or HIV-1/2, and clinically significant heart disease.
During phase A (n = 30), a single 600 mg prulifloxacin tablet was administered to fasting subjects with normal renal function, as well as to patients with mild, moderate and severe renal impairment. During phase B (n = 42), subjects with normal renal function and patients with mild and moderate renal impairment were treated, in fasting conditions, with a single 600 mg prulifloxacin tablet once daily for 7 days, while patients with severe renal impairment were treated with 300 mg prulifloxacin (half a tablet) once daily for 7 days. Categorization by group is shown in Table 1 and patient allocation is shown in Fig. 1. Demographic and anthropometric data for phases A and B are summarized in Table 2.

Patient allocation in phases A and B of the study
The treatment schedules to be used during phase B of the trial were established during the interim analysis through simulations from a population PK model built using the data obtained from phase A of the trial and from 10 previous clinical studies on prulifloxacin (Attkins et al., population PK report 2013, personal communication). The aim of simulations was to determine a dosing regimen for each of the renal impairment groups, such that the overall exposure (AUC) and the average concentration (Cmax) of ulifloxacin within the dosing range were comparable with those in subjects with normal renal function, and therefore associated with efficacy and safety. For this reason, we decided to maintain AUC and Cmax values within a twofold increase with respect to those observed in subjects with normal renal function when daily prulifloxacin 600 mg is administered in a multiple-dosage regimen.
The twofold threshold was considered as a safe margin on the basis of a previous clinical study where prulifloxacin, administered at a dose of 600 mg twice daily (every 12 h), was well tolerated (Angelini S.p.A. unpublished data).
2.2 Pharmacokinetic Analysis
During the study, blood samples were collected at given timepoints in both phase A and phase B (day 7), starting from predose up to 96 h after dosing. In addition, trough samples were collected daily in phase B from days 1–6. Urine samples were collected in phase B (day 7) starting from predose to 24 h after dosing.
Plasma and urine concentration of ulifloxacin was determined by a fully validated liquid chromatography method coupled to tandem mass spectrometry (LC-MS/MS), as already described [11]. The method validation was performed in agreement with internationally accepted standards as outlined in the applicable US FDA and European Medicines Agency (EMA) guidelines. Good Laboratory Practice (GLP) and Good Clinical Practice (GCP) principles were also applied. A calibration range of 0.00890–4.45 µg/mL was validated for the quantification of ulifloxacin in human plasma, while the analytical method for the quantification of ulifloxacin in human urine was validated over a range of 0.6832–349.9 µg/mL. PK analysis was performed using Phoenix® WinNonlin® 6.2 software (Pharsight Corporation, St Louis, MO, USA) and all the PK data were presented using the PK population.
2.3 Statistical Analysis
Statistical analysis was performed using SAS® version 9.1.3 software (SAS Institute Inc., Cary, NC, USA) or the newest version using the PK population. The statistical tests were two-sided and were performed at the 5% level of significance (if applicable).
Confidence intervals (CIs) and statistical tests were of an exploratory nature. Comparisons of plasma PK parameters were performed using analysis of variance (ANOVA), and the data were transformed using the natural logarithm prior to analysis.
Estimate statements were constructed in order to compare each renally impaired group with the healthy subjects group. The group difference (for each comparison) and the associated 95% CI estimated from the ANOVA analysis was back-transformed to obtain the estimated ratio of geometric means between the groups and the 95% CI for this ratio. In addition, tmax and tmax,ss were analysed in order to estimate the median difference and 95% CI for differences between each renally impaired group and the healthy subjects group. The 95% CI was calculated according to Hodges–Lehmann. The relationship between the individual PK parameters and renal function (eGFR and sGFR) was investigated using the linear regression analysis.
2.4 Safety Assessment
The complete safety evaluation included adverse events/severe adverse events (AEs/SAEs), vital signs, physical examination, ECG, clinical laboratory.
3 Results
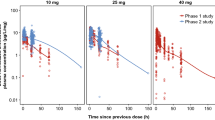
This study revealed a different increase of ulifloxacin concentration among the various renal function groups. As expected, ulifloxacin increase was observed mainly in the severe renal impairment group, with exposure increasing as renal function decreased.
During the single dose phase of the study, the reduced drug clearance led to a statistically significant increase in AUC in the severe renal impairment group, and in t½ in the moderate and severe renal impairment groups, compared with the normal renal function group. Cmax and tmax were consistent across all groups.
Linear regression analysis showed a linear relationship between eGFR and ulifloxacin exposure, with the AUC increasing as renal function decreased.
During the multiple-dose phase of the study, steady state was reached by day 3 for all groups. There was no evidence for accumulation of ulifloxacin upon multiple dosing as ulifloxacin Cmax,ss and AUCτ,ss were similar to the Cmax and AUC∞ observed during the single-dose phase of the study.
No statistically significant differences between the normal renal function group and the groups with renal impairment were shown by analysis of Cmax,ss and tmax,ss.
The AUCτ,ss increased in the groups with decreased renal function. The dose adjustment applied in the severe renal impairment group led to ulifloxacin exposure falling within the same range of the mild and moderate renal impairment groups. Also in this case, the linear regression analysis showed that there was a linear relationship between sGFR and ulifloxacin exposure, with AUC increasing as renal function decreased.
PK parameters are summarized descriptively in Table 3 for phases A and B.
The geometric mean amount of ulifloxacin excreted in urine over a 24-h dosing period was similar for both the normal function and mild renal impairment groups, but was lower for the moderate and severe renal impairment groups. Similarly, the geometric mean percentage of ulifloxacin excreted in urine decreased with renal function once the level of impairment became moderate in severity.
Linear regression analysis revealed a clear linear relationship between sGFR and renal clearance at steady state, with renal clearance decreasing as sGFR decreased. In Table 4, urine PK parameters are shown.
With regard to safety, three SAEs were reported (one event per patient). Two SAEs were classified as severe in intensity: one patient was diagnosed with chest pain and hospitalized, while another patient was hospitalized for spinal discus prolapse in the L4–L5 lumbar region. One SAE was classified as mild in intensity: one patient underwent a total left knee replacement. All SAEs, which occurred during the washout period between phase A and phase B, were judged by the investigator not to have a causal relationship with the study drug.
AEs reported during the clinical study have been stratified by degree of renal impairment and are presented in Table 5. Overall, the most commonly reported AE was headache (six events), reported in both phase A and phase B of the clinical study; all events were mild in intensity and, according to the investigator, only one was considered to have a possible causal relationship with the study drug.
No clinically significant clinical laboratory parameters, vital signs, 12-lead ECG or physical examination results were highlighted.
4 Discussion
As expected, exposure to ulifloxacin increased as renal function decreased due to a reduction in ulifloxacin clearance. Most importantly, it was found that ulifloxacin PK were significantly altered only in patients with severe renal impairment, indicating the need for dosage adjustment for this group of patients. After 7 days of drug administration with the adjusted dosage (patients with severe renal impairment were treated with a 300 mg dose), the Cmax,ss and AUCt,ss of patients with renal impairment were as shown in Table 6.
With regard to safety, as foreseen in the single-dose phase of the study, drug plasma concentrations did not fall within a warning range. Hence, there was no over concern for patients with borderline GFR values between moderate and severe renal function impairment, and no specific AEs were reported, further supporting the safety of prulifloxacin.
In addition, it should be mentioned that, despite the exploratory nature of the study, the clinical advice for dosage correction derived from the new PK data collected in renally impaired patients remains a reliable point in translation of this PK study.
It would have also been interesting to discuss the impact of demographics across the different renal function groups, however this was not in the intended scope of the study due to its exploratory nature.
The target population was selected according to the EMA guideline CHMP/EWP/225/02, dated 23 June 2004 [12], on the evaluation of the PK of medicinal products in patients with decreased renal function. During the course of the trial, a new EMA guideline (EMA/83874/2014, dated 20 February 2014 [13]) was released on this matter; however, this new guideline did not have any impact on the study results despite the sGFR measurement differing slightly for 10 patients when compared with the Cockroft–Gault method, and would have consequently resulted in some subjects changing group as the Cockroft–Gault method slightly overestimated the GFR. This could have been a potential deviation from the standard guidelines proposed for this type of study design; however, on the basis of the following considerations, it was concluded that there was no significant impact on the study:
-
(i)
GFR overestimation did not change the patient’s assigned drug dose, except for one patient who was treated with prulifloxacin 600 mg instead of 300 mg (borderline GFR between moderate and severe renal impairment).
-
(ii)
The Cockroft–Gault method is commonly used in clinical practice, and consequently its choice for grouping of subjects was considered more close-fitting to real standard of care.
5 Conclusions
To the best of our knowledge, ulifloxacin PK have been assessed for the first time in patients with different degrees of renal impairment through a clinical trial based on a specific study design. The new PK data allowed the definition of the suitable dose regimen to be applied in clinical practice in this particular population. The results showed a clear cut-off for severely renally impaired patients, which should be treated with half a tablet of prulifloxacin (corresponding to 300 mg), while patients with normal or mild-to-moderate renal impairment should be administered the usual prulifloxacin 600 mg dosage.
This study clearly shows that prulifloxacin is a safe quinolone and is well tolerated in subjects with normal renal function as well as patients with renal impairment, requiring a minimal dosage adjustment only in patients with severe renal impairment.
References
Segawa J, Kitano M, Kazuno K, et al. Studies on pyridonecarboxylic acids. 1. Synthesis and antibacterial evaluation of 7-substituted-6-halo-4-oxo-4H-[1,3]thiazeto[3,2-a]quinoline-3- carboxylic acids. J Med Chem. 1992;35:4727–38.
Blasi F, Schaberg T, Centanni S, Del Vecchio A, Rosignoli MT, Dionisio P. Prulifloxacin versus levofloxacin in the treatment of severe COPD patients with acute exacerbations of chronic bronchitis. Pulm Pharmacol Ther. 2013;26:609–16.
Passali D, Crisanti A, Bellussi LM. Role of prulifloxacin in the treatment of acute rhinosinusitis. Infez Med. 2015;23:301–6.
Keam SJ, Perry CM. Prulifloxacin. Drugs. 2004;64:2221–34.
Tougou K, Nakamura A, Watanabe S, Okuyama Y, Morino A. Paraoxonase has a major role in the hydrolysis of prulifloxacin (NM441), a prodrug of a new antibacterial agent. Drug Metab Dispos. 1998;26:355–9.
Rafailidis PI, Polyzos KA, Sgouros K, Falagas ME. Prulifloxacin: a review focusing on its use beyond respiratory and urinary tract infections. Int J Antimicrob Agents. 2011;37:283–90.
Picollo R, Brion N, Gualano V, et al. Pharmacokinetics and tolerability of prulifloxacin after single oral administration. Arzneimittelforschung. 2003;53:201–5.
Nakashima M, Uematsu T, Kosuge K, et al. Pharmacokinetics and safety of NM441, a new quinolone, in healthy male volunteers. J Clin Pharmacol. 1994;34:930–7.
Foxman B. Epidemiology of urinary tract infections: incidence, morbidity, and economic costs. Am J Med. 2002;113:5S–13S.
Vestbo J, Hurd SS, Agusti AG, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2013;187:347–65.
De Benedetto M, Passali D, Tomacelli G, et al. Penetration of prulifloxacin into sinus mucosa of patients undergoing paranasal sinus elective endoscopic surgery. J Chemother. 2012;24:26–31.
EMA guideline CHMP/EWP/225/02, dated June 23, 2004. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003123.pdf.
EMA guideline EMA/83874/2014, dated February 20, 2014. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/02/WC500162133.pdf.
Acknowledgements
Medical editing assistance was provided by Content Ed Net, with the helpful assistance of Amalia Forte, PhD.
Author information
Authors and Affiliations
Contributions
VT, PC, RP and GDL contributed to study conception and design; PC, RP and GDL were involved in the review of data analysis and interpretation; VT, PC, MA and GDL contributed to manuscript preparation; and ADV, RP, AC, FG and ST contributed to manuscript editing. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Funding
The study was funded by Angelini S.p.A.
Conflict of interest
Valeria Tellone, Paola Coppola, Marco Ammendola, Giorgio Di Loreto, Rossella Picollo, Alessandra Del Vecchio, Alessandro Comandini, Fabio Garofolo and Serena Tongiani are employees of Angelini S.p.A. Paola Coppola and Marco Ammendola were employees of Angelini S.p.A. at the time of clinical trial conduction and writing of this manuscript.
Ethics approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Consent to participate
Informed consent was obtained from all individual participants included in this study.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Tellone, V., Coppola, P., Ammendola, M. et al. New Insights on the Pharmacokinetics of Ulifloxacin After Administration of Prulifloxacin in Patients with Mild, Moderate and Severe Renal Impairment. Drugs R D 18, 237–245 (2018). https://doi.org/10.1007/s40268-018-0245-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-018-0245-9