
Abstract
Epidemiological, clinical, and experimental evidence has accumulated during the last decades suggesting that high-density lipoproteins (HDLs) may protect from atherosclerosis and its clinical consequences. However, more than 55 years after the first description of the link between HDL and heart attacks, many facets of the biochemistry, function, and clinical significance of HDL remain enigmatic. This applies particularly to the completely unexpected results that became available from some recent clinical trials of nicotinic acid and of inhibitors of cholesteryl ester transfer protein (CETP). The concept that raising HDL cholesterol by pharmacological means would decrease the risk of vascular disease has therefore been challenged.
1 Early History, the High-Density Lipoprotein (HDL) Hypothesis and the Enigma
In 1988, Paul Williams at the Donner Laboratory, University of California, Berkeley, USA, recovered a dusty stack of punch cards. The cards contained data from the Livermore cohort study that had been started by John Gofman (Fig. 1) between 1954 and 1956. Along with Howard Eder and colleagues [1], Gofman was among the first to recognize that low concentrations of high-density lipoproteins (HDLs) are associated with a high risk of coronary disease, and he wished to further substantiate the findings in a prospective fashion [2]. In 1966, Gofman published the first results of his study [3], but these became buried into near oblivion and it took more than a decade before the HDL hypothesis was ‘re-invented’ [4].

John Gofman in front of an analytical ultracentrifuge at the Donner Laboratory, University of California, Berkeley, USA (approximately 1948)
Williams restored Gofman’s data using an old-fashioned punch card machine and succeeded in retrieving the clinical history of more than 97 % of the 1,905 men who were studied originally. Over an observation period of 29 years, a total of 363 incidents due to coronary heart disease had occurred and indeed, both HDL2 and HDL3 turned out to be inversely associated with coronary risk [5]. However, today, more than 55 years after the first description of the link between HDL and heart attacks, many facets of the biochemistry, function, and clinical significance of HDL remain enigmatic. This applies particularly to the completely unexpected, basically neutral, results that recently became available from clinical trials. We here embody our view of why trialists have unavailingly spent a fortune on the hypothesis that merely raising the steady-state concentrations of HDL cholesterol (HDL-C) would yield clinical benefit.
2 Nicotinic Acid and the GPR109A Receptor
Of the conventional lipid drugs, it is primarily the fibrates and nicotinic acid that raise HDL-C, and both of these agents reduce cardiovascular endpoints [6, 7]. However, fibrates and nicotinic acid also reduce the levels of triglycerides and low-density lipoprotein cholesterol (LDL-C), and it is therefore not possible to draw any definite conclusions on the clinical benefit attributable to the rise in HDL-C alone. The evaluation of nicotinic acid has also grown more complex than originally conceived, since commonly accepted assumptions on its mechanism of action have been proved wrong. It was thought that nicotinic acid inhibits lipolysis by binding to the G protein-coupled receptor GPR109A, the decreased availability of free fatty acids consequentially limiting the formation of very low-density lipoproteins (VLDLs) and LDL. Recently, Lauring and colleagues [8], showed that, while GPR109A is involved in the inhibition of lipolysis (and the occurrence of the well known troublesome flushes), it does not mediate the effects on lipoproteins. Beyond its lipid-modifying effects, nicotinic acid does have anti-inflammatory actions that are mediated at least in part by GPR109A [9, 10].
3 Two Trials of Nicotinic Acid: AIM-HIGH and HPS2-THRIVE
Recent efforts to re-evaluate nicotinic acid had been prompted by the results of the Coronary Drug Project and other smaller studies. In the Coronary Drug Project, nicotinic acid decreased the incidence rate of myocardial infarction, but no reduction in mortality was observed within the study period. However, after 15 years, the overall mortality in the group that had originally received nicotinic acid was significantly lower than in the placebo group [11]. A meta-analysis of randomized trials with nicotinic acid (usually without concomitant statin dose) reported a reduction in coronary events by 25 %, stroke by 26 %, and any cardiovascular events by 27 % by nicotinic acid [7]. Another meta-analysis of 11 studies, mainly of secondary prevention and also including the AIM-HIGH also showed a reduction in cardiovascular endpoints by 34 % and coronary endpoints by 25 % by nicotinic acid [12].
In view of the complex and pleiotropic actions of nicotinic acid, it would be illogical to consider the ‘inefficiency’ of nicotinic acid in the recently completed AIM-HIGH trial as an argument against the clinical value of modifying HDL-C in general. In the AIM-HIGH trial, extended-release niacin (1,500 daily to 2,000 mg) was compared with a small and non-lipid-efficacious dose of nicotinic acid in 3,414 patients with cardiovascular disease who received simvastatin with or without ezetimibe to titrate LDL-C to 40 to 80 mg/dl (3.1–7.2 mmol/l). The study was stopped early after 3 years, because an interim analysis indicated that continuation would have been unlikely to yield a difference between the two arms of the study. At the end of treatment, event rates were similar in the two groups. The LDL-C concentration had further decreased by 12 % in the niacin group and by 5.5 % in the placebo group, corresponding to an absolute difference in LDL-C during follow-up of approximately 5 mg/dl (0.13 mmol/l) between the two treatment arms. HDL-C only differed by 7 mg/dl (0.18 mmol/l). These differences were probably too small to have a significant effect on cardiovascular endpoints [13]. Moreover, it was speculated that the small amounts of nicotinic acid given in the control group (for provoking flush and to facilitate blinding) may have already elicited positive cardiovascular effects (unrelated to lipids). Thus the AIM-HIGH trial was in reality not a placebo-controlled trial but rather a comparison of a low-dose with a high-dose of nicotinic acid where the ‘placebo’ group might have received a dose of nicotinic acid sufficiently large enough to affect the well-being of the patient, independent of lipoproteins, including HDL.
The Heart Protection Study 2-Treatment of HDL to Reduce the Incidence of Vascular Events (HPS2-THRIVE) enrolled 25,673 men and women with vascular disease (over 8,000 of these are from the UK with close to 11,000 from China, and 6,500 from Scandinavia). In that study, the combination of a statin (plus optionally ezetimibe) and extended-release niacin in a fixed combination with laropiprant, an anti-flushing agent (Tredaptive®), was compared with statin (plus optionally ezetimibe) therapy alone. The preliminary HPS2-THRIVE results made public on 20 December 2012 show that, when added to an effective statin-based treatment, the combination of extended-release niacin and laropiprant did not produce clinically meaningful reductions in the rate of major vascular events (such as heart attacks, strokes, or revascularizations) after a median follow-up of 3.9 years. It should be noted that baseline lipoprotein concentrations in this trial were in a range that would not have prompted clinicians to commence additional lipid-lowering drugs: total cholesterol 128 mg/dl (3.32 mmol/l), LDL-C 63 mg/dl (1.64 mmol/l), HDL-C 44 mg/dl (1.14 mmol/l), triglycerides 125 mg/dl (1.43 mmol/l).
There was also a statistically significant increase in the incidence of some types of non-fatal serious adverse events in the group that received niacin/laropiprant. While this indication of harm needs further scrutiny in relation to the known issues with niacin treatment, once the final and detailed results are available, the question whether the failure of HPS2-THRIVE was due to any off-target effects of laropiprant with niacin will ultimately be hard to resolve. It would further be interesting to see if patients with very low HDL-C had any clinical benefit. In our opinion, the results of HPS2-THRIVE most likely reflect that (i) it is and will be hard anyway to demonstrate additional improvement in outcomes once LDL-C is well controlled and that (ii) HDLs might lose their cardio-protective functions in patients who have advanced atherosclerosis (see below). The results of HPS2-THRIVE available as of this writing bear two other incidental, but potentially interesting, aspects. First, 13,542 patients received monotherapy with simvastatin and 12,131 patients required combination therapy simvastatin/ezetimibe. The combination therapy lowered LDL-C by 4 mg/dl (0.10 mmol/l) over monotherapy. This can be expected to result in a relative reduction of the incidence rate of major vascular events by no more than 2 %. [14] Actually, 14 % of the study participants had an event with simvastatin alone, compared with 12.8 % with simvastatin/ezetimibe (relative risk reduction 8.6 %, p = 0.008 by χ2 test), suggesting that the lowering of LDL-C attributable to ezetimibe is biologically at least equivalent to that produced by statins [14]. Second, there was a statistically significant interaction between LDL-C at baseline and the effect of niacin/laropiprant, suggesting that niacin/laropiprant might have been effective if starting LDL-C had been higher.
4 Inhibitors of the Cholesteryl Ester Transfer Protein (CETP)
Far more effective in raising HDL-C than either nicotinic acid or fibrates are inhibitors of the cholesteryl ester transfer protein (CETP). CETP is a hydrophobic glycoprotein that mediates the transfer of cholesteryl esters from HDL to VLDL and LDL in exchange for triglycerides [15]. As a consequence, CETP decreases HDL-C, and cholesteryl esters delivered to LDL are subsequently and mainly passed to the liver [16]. This illustrates that not only HDL but also CETP and LDL may be important links in the transport of cholesterol from peripheral tissues to the liver (reverse cholesterol transport). At least theoretically, inhibiting CETP may thus compromise rather than accelerate reverse cholesterol transport despite raising HDL-C. In recent years, four CETP inhibitors have been tested clinically (torcetrapib, dalcetrapib, anacetrapib [17, 18], and evacetrapib [19]). The development of torcetrapib [20] was terminated in 2006 because it significantly increased both total mortality and cardiovascular mortality in a randomized phase III clinical trial (ILLUMINATE [Investigation of Lipid Level Management to Understand Its Impact in Atherosclerotic Events]) [20]. In smaller studies, torcetrapib consistently also had no protective effects on surrogate cardiovascular endpoints [21–23]. However, doubts about CETP inhibition as a treatment approach were rapidly dispelled after it turned out that torcetrapib stimulated the formation of aldosterone and raised blood pressure [24, 25], independent of the lipid effects. Unlike torcetrapib, dalcetrapib has so far been considered to leave blood pressure unaffected, and there was hope that dalcetrapib would reduce clinical events. Faint skepticism emerged after the results of two small studies primarily addressing the safety of dalcetrapib became available: dalcetrapib neither had a discernible effect on the function of the vascular endothelium [26] nor had it compelling effects on structural features (magnetic resonance imaging [MRI]) and the inflammatory activity (positron emission tomography/computed tomography [PET/CT]) of the carotid arteries [27]. A direct correspondence between studies of these surrogate vascular endpoints and clinical outcomes might not exist. However, the biggest disappointment then was that the endpoint study Dal-OUTCOMES [16, 28] was terminated prematurely in May 2012. Patients (n = 15,871) who had experienced an acute coronary syndrome received either dalcetrapib (600 mg daily) or placebo in addition to standard therapy, which produced a mean LDL-C at baseline of 76 mg/dl (1.97 mmol/l). As expected, dalcetrapib increased HDL-C by 31–40 %, with virtually no effect on LDL-C. After a median follow-up of 31 months, 71 % of the projected primary endpoints had occurred, and event rates were not different between dalcetrapib and placebo. Interestingly, the median C-reactive protein was, on average, 0.2 mg/l (18 %) higher and the mean systolic blood pressure was 0.6 mmHg higher in the dalcetrapib arm than in the placebo arm.
A number of reasons for the lack of a clinical effect of dalcetrapib are conceivable:
-
1.
The increase in HDL-C (31–40 %) produced by the drug may have been too small. On the other hand, increases in HDL-C exceeding this magnitude may not entail any further clinical advantage. The relationship between HDL-C and risk is not linear, and, beyond 60 mg/dl (1.5 mmol/l), increases may not translate into further risk reduction (Fig. 2) [29]. Yet, coronary risk could even increase again [30].
Fig. 2 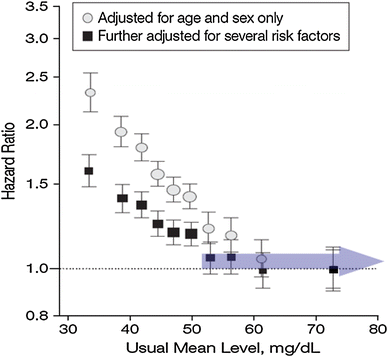
High-density lipoprotein cholesterol and coronary heart disease [29]
-
2.
Fundamental differences may exist between dalcetrapib and other inhibitors of CETP. Anacetrapib and torcetrapib seem to occupy a similar binding site on CETP, which is different from the binding site of dalcetrapib [31]. Dalcetrapib, but neither torcetrapib nor anacetrapib induce specific conformational changes in the structure of CETP so that antibodies against specific epitopes of CETP cannot bind any more. In another study, dalcetrapib significantly more effectively enhanced fecal cholesterol than torcetrapib [32].
-
3.
In addition to their incriminated role in reverse cholesterol transport, HDLs may possess other cardio-protective properties. These include anti-oxidant effects, improvement of endothelial nitric oxide production, reduction of leukocyte adhesion, reduced apoptosis of endothelial cells, inhibition of platelet aggregation, and blood coagulation. HDL isolated from patients with very high-risk or overt coronary artery disease (CAD) may have lost these functions [33, 34]. Of course, increasing the concentration of such ‘dysfunctional’ HDL would predictably not be very useful. In the patients with an acute coronary syndrome, as they participated in Dal-OUTCOMES, coronary heart disease could have been advanced that far so that HDL may have lost their protective effect. Two very recent studies support this possibility [35–37] Angeloni et al. [36] found no (or even a trend of a positive) association of HDL-C with cardiovascular events and death in patients with CAD undergoing coronary artery bypass graft surgery. In the LURIC (Ludwigshafen Risk and Cardiovascular Health) study, we demonstrated a strong inverse relationship between HDL-C and cardiovascular mortality in subjects without angiographic CAD, but not once CAD was established. We confirmed this interaction of HDL-C with CAD in determining adverse outcomes in two independent cohorts and by a meta-analysis of all three cohorts (n = 12,292) [35].
If it is true that HDL become dysfunctional in CAD patients, HDL-raising agents should in the future be tested preferably in persons with low HDL, but without overt vascular disease, which would make clinical trials even more demanding than they have been so far.
-
4.
The approach to increase HDL-C by inhibiting CETP could be wrong. The measurable concentration of HDL-C is influenced by many metabolic processes such as the production rate of the apolipoproteins of HDL, and the activities of membrane cholesterol transporters (ABCA1 and ABCG1), lipid transfer proteins (CETP, phospholipid transfer protein), and enzymes (endothelial lipase, hepatic lipase, lecithin cholesterol acyltransferase [LCAT]) (Fig. 3). Epidemiological findings suggest that high CETP increases and inhibition of CETP reduces the risk of atherosclerosis [38]. Further support for this assumption comes from observations that CETP activity was positively related to subsequent cardiovascular events [39, 40]. However, other studies have paradoxically observed the opposite when measuring CETP activity [41–44] or concentration [45]. Experimental studies, especially in rabbits (which have high CETP activity naturally), show atherosclerosis following CETP inhibition [46, 47]. In mice (which lack CETP naturally), human CETP was introduced as a transgene, and the effects of CETP were studied. Some authors reported positive effects [48] while others reported negative effects [49, 50]. In contrast, in both animal experiments and humans, the parenteral administration of (presumably functional) HDL consistently has had anti-atherosclerotic effects so far [51–54] This could indicate that raising HDL by enhancing its production might ultimately turn out to be preferable over blocking processing with inhibitors of CETP. Further, CETP inhibitors have been reported to produce increases in blood pressure or C-reactive protein. Because these effects are apparently not limited to torcetrapib [18, 20, 55], they may represent a class effect or simply relate to lowering CETP activity and this may offset other cardiovascular benefit.
Fig. 3 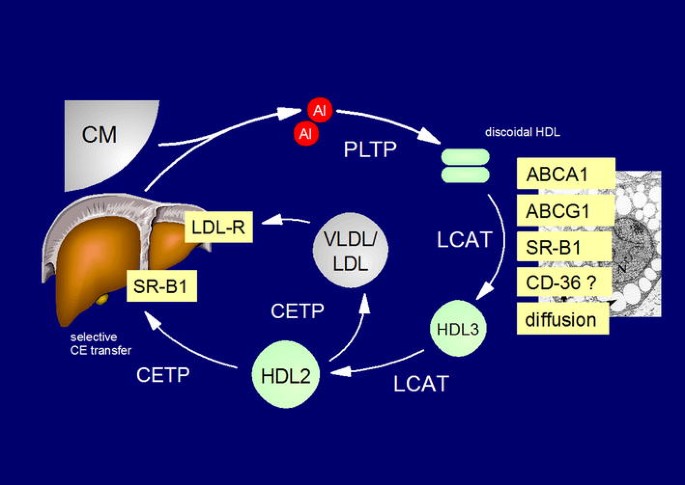
Reverse cholesterol transport. ABCA1 ATP-binding cassette transporter A1, ABCG1 ATP-binding cassette transporter G1, AI apolipoprotein AI, CD-36 cluster of differentiation 36, CETP cholesteryl ester transfer protein, CM chylomicrons, HDLs high-density lipoproteins, LCAT lecithin cholesterol-acyl transferase, LDLs low-density lipoproteins, LDL-R LDL receptor, PLTP phospholipid transfer protein, SR-B1 scavenger receptor B1, thrombospondin receptor, VLDL very low density lipoproteins,
-
5.
The approach to increase HDL-C by any pharmacological intervention could be wrong. The inverse correlation between HDL-C and coronary events may merely reflect ‘reverse causality‘. It is in line with this idea that Mendelian randomization studies argue against a causal role of low HDL in vascular disease. [56] Thus, pre-existing, yet clinically asymptomatic vascular disease and associated subclinical inflammation could alter the composition of HDL and lower the HDL-C. ‘Acute phase HDL’ are characterized by enrichment of serum amyloid A, secretory phospholipase A2 (sPLA2-IIa), ceruloplasmin, or apoCIII, and a lower percentage of apo AI, paraoxonase, and clusterin [57, 58]. Whether the function of HDL as an acceptor of cellular cholesterol is impaired in the acute phase is not entirely clear [59].


Because LDL-C, apolipoprotein B, and triglycerides remained virtually unaffected in the Dal-OUTCOMES study, the study may indeed provide clues to the utility of isolated manipulation of HDL-C. However, as outlined above there are many other explanations for the neutral results of the trial. Two CETP inhibitors, anacetrapib and evacetrapib, are still under evaluation. They increase HDL-C by 100 % or more. Beyond that, they also lower LDL-C. Based on the latter effect, they may be more effective in reducing cardiovascular events than dalcetrapib.
5 Conclusions
The central question as to whether a therapeutic increase in HDL-C in general would result in relevant cardiovascular risk reduction may thus stay an enigma for a long time still. We believe that researchers will have to step back to understand the functions of HDL and its subfractions and the dynamics of HDL metabolism before other drugs targeting HDL components can successfully be developed.
References
Barr DP, Russ EM, Eder HA. Protein–lipid relationships in human plasma. II. In atherosclerosis and related conditions. Am J Med. 1951;11:480–93.
Gofman JW. The natural of the relationship of disturbance in blood lipid transport with the evolution of clinical coronary heart disease. Trans Am Coll Cardiol. 1955;4:198–220.
Gofman JW, Young W, Tandy R. Ischemic heart disease, atherosclerosis, and longevity. Circulation. 1966;34:679–97.
Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR. High density lipoprotein as a protective factor against coronary heart disease. Am J Med. 1977;62:707–14.
Williams PT, Feldman DE. Prospective study of coronary heart disease vs. HDL2, HDL3, and other lipoproteins in Gofman’s Livermore cohort. Atherosclerosis. 2011;214:196–202.
Jun M, Foote C, Lv J, Neal B, Patel A, Nicholls SJ, Grobbee DE, Cass A, Chalmers J, Perkovic V. Effects of fibrates on cardiovascular outcomes: a systematic review and meta-analysis. Lancet. 2010; 375:1875–84.
Bruckert E, Labreuche J, Amarenco P. Meta-analysis of the effect of nicotinic acid alone or in combination on cardiovascular events and atherosclerosis. Atherosclerosis. 2010;210:353–61.
Lauring B, Taggart AK, Tata JR, Dunbar R, Caro L, Cheng K, Chin J, Colletti SL, Cote J, Khalilieh S, Liu J, Luo WL, Maclean AA, Peterson LB, Polis AB, Sirah W, Wu TJ, Liu X, Jin L, Wu K, Boatman PD, Semple G, Behan DP, Connolly DT, Lai E, Wagner JA, Wright SD, Cuffie C, Mitchel YB, Rader DJ, Paolini JF, Waters MG, Plump A. Niacin lipid efficacy is independent of both the niacin receptor GPR109a and free fatty acid suppression. Sci Transl Med. 2012;4:148ra115.
Lukasova M, Hanson J, Tunaru S, Offermanns S. Nicotinic acid (niacin): new lipid-independent mechanisms of action and therapeutic potentials. Trends Pharmacol Sci. 2011;32:700–7.
Offermanns S. It ain’t over ’til the fat lady sings. Sci Transl Med. 2012;4:148fs130.
Canner PL, Berge KG, Wenger WK, Stamler J, Friedman L, Prineas RJ, Friedewald W, Group for the Coronary Drug Project Group. Fifteen year mortality in Coronary Drug Project patients: long-term benefit with niacin. J Am Coll Cardiol. 1986;8:1245–55.
Lavigne PM, Karas RH. The current state of niacin in cardiovascular disease prevention: a systematic review and meta-regression. J Am Coll Cardiol. 2013;61:440–6.
Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes-Nickens P, Koprowicz K, McBride R, Teo K, Weintraub W. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365:2255–2267.
Masana L, Cabre A, Plana N. HPS2-thrive results: Bad for niacin/laropiprant, good for ezetimibe? Atherosclerosis. 2013;229:449–50.
Quintao EC, Cazita PM. Lipid transfer proteins: past, present and perspectives. Atherosclerosis. 2010;209:1–9.
Schwartz CC, VandenBroek JM, Cooper PS. Lipoprotein cholesteryl ester production, transfer, and output in vivo in humans. J Lipid Res. 2004;45:1594–607.
Masson D. Anacetrapib, a cholesterol ester transfer protein (CETP) inhibitor for the treatment of atherosclerosis. Curr Opin Investig Drugs. 2009;10:980–7.
Cannon CP, Shah S, Dansky HM, Davidson M, Brinton EA, Gotto AM, Stepanavage M, Liu SX, Gibbons P, Ashraf TB, Zafarino J, Mitchel Y, Barter P. Safety of anacetrapib in patients with or at high risk for coronary heart disease. N Engl J Med. 2010;363:2406–15.
Nicholls SJ, Brewer HB, Kastelein JJ, Krueger KA, Wang MD, Shao M, Hu B, McErlean E, Nissen SE. Effects of the CETP inhibitor evacetrapib administered as monotherapy or in combination with statins on HDL and LDL cholesterol: a randomized controlled trial. JAMA. 2011;306:2099–109.
Barter PJ, Caulfield M, Eriksson M, Grundy SM, Kastelein JJ, Komajda M, Lopez-Sendon J, Mosca L, Tardif JC, Waters DD, Shear CL, Revkin JH, Buhr KA, Fisher MR, Tall AR, Brewer B. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357:2109–22.
Kastelein JJ, van Leuven SI, Burgess L, Evans GW, Kuivenhoven JA, Barter PJ, Revkin JH, Grobbee DE, Riley WA, Shear CL, Duggan WT, Bots ML. Effect of torcetrapib on carotid atherosclerosis in familial hypercholesterolemia. N Engl J Med. 2007;356:1620–30.
Bots ML, Visseren FL, Evans GW, Riley WA, Revkin JH, Tegeler CH, Shear CL, Duggan WT, Vicari RM, Grobbee DE, Kastelein JJ. Torcetrapib and carotid intima-media thickness in mixed dyslipidaemia (RADIANCE 2 study): a randomised, double-blind trial. Lancet. 2007;370:153–60.
Nissen SE, Tardif JC, Nicholls SJ, Revkin JH, Shear CL, Duggan WT, Ruzyllo W, Bachinsky WB, Lasala GP, Tuzcu EM. Effect of torcetrapib on the progression of coronary atherosclerosis. N Engl J Med. 2007;356:1304–16.
Vergeer M, Bots ML, van Leuven SI, Basart DC, Sijbrands EJ, Evans GW, Grobbee DE, Visseren FL, Stalenhoef AF, Stroes ES, Kastelein JJ. Cholesteryl ester transfer protein inhibitor torcetrapib and off-target toxicity: a pooled analysis of the rating atherosclerotic disease change by imaging with a new CETP inhibitor (RADIANCE) trials. Circulation. 2008;118:2515–22.
Forrest MJ, Bloomfield D, Briscoe RJ, Brown PN, Cumiskey AM, Ehrhart J, Hershey JC, Keller WJ, Ma X, McPherson HE, Messina E, Peterson LB, Sharif-Rodriguez W, Siegl PK, Sinclair PJ, Sparrow CP, Stevenson AS, Sun SY, Tsai C, Vargas H, Walker M 3rd, West SH, White V, Woltmann RF. Torcetrapib-induced blood pressure elevation is independent of CETP inhibition and is accompanied by increased circulating levels of aldosterone. Br J Pharmacol. 2008;154:1465–73.
Luscher TF, Taddei S, Kaski JC, Jukema JW, Kallend D, Münzel T, Kastelein JJ, Deanfield JE. Vascular effects and safety of dalcetrapib in patients with or at risk of coronary heart disease: the DAL-vessel randomized clinical trial. Eur Heart J. 2012;33:857–65.
Fayad ZA, Mani V, Woodward M, Kallend D, Abt M, Burgess T, Fuster V, Ballantyne CM, Stein EA, Tardif JC, Rudd JH, Farkouh ME, Tawakol A. Safety and efficacy of dalcetrapib on atherosclerotic disease using novel non-invasive multimodality imaging (DAL-plaque): a randomised clinical trial. Lancet. 2011;378:1547–59.
Schwartz GG, Olsson AG, Ballantyne CM, Barter PJ, Holme IM, Kallend D, Leiter LA, Leitersdorf E, McMurray JJ, Shah PK, Tardif JC, Chaitman BR, Duttlinger-Maddux R, Mathieson J. Rationale and design of the DAL-outcomes trial: Efficacy and safety of dalcetrapib in patients with recent acute coronary syndrome. Am Heart J. 2009;158(896–901):e893.
Di Angelantonio E, Sarwar N, Perry P, Kaptoge S, Ray KK, Thompson A, Wood AM, Lewington S, Sattar N, Packard CJ, Collins R, Thompson SG, Danesh J. Major lipids, apolipoproteins, and risk of vascular disease. JAMA. 2009;302:1993–2000.
van der Steeg WA, Holme I, Boekholdt SM, Larsen ML, Lindahl C, Stroes ES, Tikkanen MJ, Wareham NJ, Faergeman O, Olsson AG, Pedersen TR, Khaw KT, Kastelein JJ. High-density lipoprotein cholesterol, high-density lipoprotein particle size, and apolipoprotein A-I: significance for cardiovascular risk: the IDEAL and EPIC-Norfolk studies. J Am Coll Cardiol. 2008;51:634–42.
Maugeais C, Magg C, Dernick G, Matile H, von der Mark E, Pflieger P, et al. Dalcetrapib binds to and changes the conformation of CETP in a unique manner. American Heart Association, Scientific Sessions, November 16, 2009, Poster 1092.
Niesor EJ, Okamoto H, Maugeais C, Von der Mark E, Brousse M, Blum-Kaelin D, et al. The effects of dalcetrapib on macrophage reverse cholesterol transport in a rodent model. American Heart Association Scientific Sessions, November 17, 2009, Presentation 1271.
Besler C, Heinrich K, Rohrer L, Doerries C, Riwanto M, Shih DM, Chroni A, Yonekawa K, Stein S, Schaefer N, Mueller M, Akhmedov A, Daniil G, Manes C, Templin C, Wyss C, Maier W, Tanner FC, Matter CM, Corti R, Furlong C, Lusis AJ, von Eckardstein A, Fogelman AM, Lüscher TF, Landmesser U. Mechanisms underlying adverse effects of HDL on ENOS-activating pathways in patients with coronary artery disease. J Clin Invest. 2011;121:2693–708.
Sorrentino SA, Besler C, Rohrer L, Meyer M, Heinrich K, Bahlmann FH, Mueller M, Horvath T, Doerries C, Heinemann M, Flemmer S, Markowski A, Manes C, Bahr MJ, Haller H, von Eckardstein A, Drexler H, Landmesser U. Endothelial-vasoprotective effects of high-density lipoprotein are impaired in patients with type 2 diabetes mellitus but are improved after extended-release niacin therapy. Circulation. 2010;121:110–22.
Silbernagel G, Schöttker B, Appelbaum S, Scharnagl H, Kleber ME, Grammer TB, Ritsch A, Mons U, Holleczek B, Goliasch G, Niessner A, Boehm BO, Schnabel RB, Brenner H, Blankenberg S, Landmesser U, März W. High-density lipoprotein cholesterol, coronary artery disease, and cardiovascular mortality. Eur Heart J. 2013;34:3563–71.
Angeloni E, Paneni F, Landmesser U, Benedetto U, Melina G, Luscher TF, Volpe M, Sinatra R, Cosentino F. Lack of protective role of HDL-c in patients with coronary artery disease undergoing elective coronary artery bypass grafting. Eur Heart J. 2013;34:3557–62.
Shah PK. Jekyll and Hyde of HDL: A lipoprotein with a split personality. Eur Heart J. 2013;34:3531–4.
van der Steeg WA, Kuivenhoven JA, Klerkx AH, Boekholdt SM, Hovingh GK, Kastelein JJ. Role of CETP inhibitors in the treatment of dyslipidemia. Curr Opin Lipidol. 2004;15:631–6.
Boekholdt SM, Kuivenhoven JA, Wareham NJ, Peters RJ, Jukema JW, Luben R, Bingham SA, Day NE, Kastelein JJ, Khaw KT. Plasma levels of cholesteryl ester transfer protein and the risk of future coronary artery disease in apparently healthy men and women: The prospective EPIC (European Prospective Investigation into Cancer and Nutrition)-Norfolk population study. Circulation. 2004;110:1418–23.
Curb JD, Abbott RD, Rodriguez BL, Masaki K, Chen R, Sharp DS, Tall AR. A prospective study of HDL-c and cholesteryl ester transfer protein gene mutations and the risk of coronary heart disease in the elderly. J Lipid Res. 2004;45:948–53.
Zhong S, Sharp DS, Grove JS, Bruce C, Yano K, Curb JD, Tall AR. Increased coronary heart disease in Japanese-American men with mutation in the cholesteryl ester transfer protein gene despite increased HDL levels. J Clin Invest. 1996;97:2917–23.
Borggreve SE, Hillege HL, Wolffenbuttel BH, de Jong PE, Zuurman MW, van der Steege G, van Tol A, Dullaart RP. An increased coronary risk is paradoxically associated with common cholesteryl ester transfer protein gene variations that relate to higher high-density lipoprotein cholesterol: a population-based study. J Clin Endocrinol Metab. 2006;91:3382–8.
Agerholm-Larsen B, Tybjaerg-Hansen A, Schnohr P, Steffensen R, Nordestgaard BG. Common cholesteryl ester transfer protein mutations, decreased HDL cholesterol, and possible decreased risk of ischemic heart disease: the Copenhagen City Heart Study. Circulation. 2000;102:2197–203.
Vasan RS, Pencina MJ, Robins SJ, Zachariah JP, Kaur G, D’Agostino RB, Ordovas JM. Association of circulating cholesteryl ester transfer protein activity with incidence of cardiovascular disease in the community. Circulation. 2009;120:2414–20.
Ritsch A, Scharnagl H, Eller P, Tancevski I, Duwensee K, Demetz E, Sandhofer A, Boehm BO, Winkelmann BR, Patsch JR, März W. Cholesteryl ester transfer protein and mortality in patients undergoing coronary angiography: the Ludwigshafen Risk and Cardiovascular Health study. Circulation. 2010;121:366–74.
Gaofu Q, Jun L, Xiuyun Z, Wentao L, Jie W, Jingjing L. Antibody against cholesteryl ester transfer protein (CETP) elicited by a recombinant chimeric enzyme vaccine attenuated atherosclerosis in a rabbit model. Life Sci. 2005;77:2690–702.
Morehouse LA, Sugarman ED, Bourassa PA, Sand TM, Zimetti F, Gao F, Rothblat GH, Milici AJ. Inhibition of CETP activity by torcetrapib reduces susceptibility to diet-induced atherosclerosis in New Zealand white rabbits. J Lipid Res. 2007;48:1263–72.
Harder C, Lau P, Meng A, Whitman SC, McPherson R. Cholesteryl ester transfer protein (CETP) expression protects against diet induced atherosclerosis in sr-bi deficient mice. Arterioscler Thromb Vasc Biol. 2007;27:858–64.
Plump AS, Masucci-Magoulas L, Bruce C, Bisgaier CL, Breslow JL, Tall AR. Increased atherosclerosis in APOE and LDL receptor gene knock-out mice as a result of human cholesteryl ester transfer protein transgene expression. Arterioscler Thromb Vasc Biol. 1999;19:1105–10.
Van Eck M, Ye D, Hildebrand RB, Kar Kruijt J, de Haan W, Hoekstra M, Rensen PC, Ehnholm C, Jauhiainen M, Van Berkel TJ. Important role for bone marrow-derived cholesteryl ester transfer protein in lipoprotein cholesterol redistribution and atherosclerotic lesion development in LDL receptor knockout mice. Circ Res. 2007;100:678–85.
Nissen SE, Tsunoda T, Tuzcu EM, Schoenhagen P, Cooper CJ, Yasin M, Eaton GM, Lauer MA, Sheldon WS, Grines CL, Halpern S, Crowe T, Blankenship JC, Kerensky R. Effect of recombinant APOA-I milano on coronary atherosclerosis in patients with acute coronary syndromes: a randomized controlled trial. JAMA. 2003;290:2292–300.
Nicholls SJ, Cutri B, Worthley SG, Kee P, Rye KA, Bao S, Barter PJ. Impact of short-term administration of high-density lipoproteins and atorvastatin on atherosclerosis in rabbits. Arterioscler Thromb Vasc Biol. 2005;25:2416–21.
Tardif JC, Gregoire J, L’Allier PL, Ibrahim R, Lesperance J, Heinonen TM, Kouz S, Berry C, Basser R, Lavoie MA, Guertin MC, Rodes-Cabau J. Effects of reconstituted high-density lipoprotein infusions on coronary atherosclerosis: a randomized controlled trial. JAMA. 2007;297:1675–82.
Sacks FM, Rudel LL, Conner A, Akeefe H, Kostner G, Baki T, Rothblat G, de la Llera-Moya M, Asztalos B, Perlman T, Zheng C, Alaupovic P, Maltais JA, Brewer HB. Selective delipidation of plasma HDL enhances reverse cholesterol transport in vivo. J Lipid Res. 2009;50:894–907.
Schwartz GG, Olsson AG, Abt M, Ballantyne CM, Barter PJ, Brumm J, Chaitman BR, Holme IM, Kallend D, Leiter LA, Leitersdorf E, McMurray JJ, Mundl H, Nicholls SJ, Shah PK, Tardif JC, Wright RS. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med. 2012;367:2089–99.
Voight BF, Peloso GM, Orho-Melander M, Frikke-Schmidt R, Barbalic M, Jensen MK, Hindy G, Holm H, Ding EL, Johnson T, Schunkert H, Samani NJ, Clarke R, Hopewell JC, Thompson JF, Li M, Thorleifsson G, Newton-Cheh C, Musunuru K, Pirruccello JP, Saleheen D, Chen L, Stewart A, Schillert A, Thorsteinsdottir U, Thorgeirsson G, Anand S, Engert JC, Morgan T, Spertus J, Stoll M, Berger K, Martinelli N, Girelli D, McKeown PP, Patterson CC, Epstein SE, Devaney J, Burnett MS, Mooser V, Ripatti S, Surakka I, Nieminen MS, Sinisalo J, Lokki ML, Perola M, Havulinna A, de Faire U, Gigante B, Ingelsson E, Zeller T, Wild P, de Bakker PI, Klungel OH, Maitland-van der Zee AH, Peters BJ, de Boer A, Grobbee DE, Kamphuisen PW, Deneer VH, Elbers CC, Onland-Moret NC, Hofker MH, Wijmenga C, Verschuren WM, Boer JM, van der Schouw YT, Rasheed A, Frossard P, Demissie S, Willer C, Do R, Ordovas JM, Abecasis GR, Boehnke M, Mohlke KL, Daly MJ, Guiducci C, Burtt NP, Surti A, Gonzalez E, Purcell S, Gabriel S, Marrugat J, Peden J, Erdmann J, Diemert P, Willenborg C, Konig IR, Fischer M, Hengstenberg C, Ziegler A, Buysschaert I, Lambrechts D, Van de Werf F, Fox KA, El Mokhtari NE, Rubin D, Schrezenmeir J, Schreiber S, Schafer A, Danesh J, Blankenberg S, Roberts R, McPherson R, Watkins H, Hall AS, Overvad K, Rimm E, Boerwinkle E, Tybjaerg-Hansen A, Cupples LA, Reilly MP, Melander O, Mannucci PM, Ardissino D, Siscovick D, Elosua R, Stefansson K, O’Donnell CJ, Salomaa V, Rader DJ, Peltonen L, Schwartz SM, Altshuler D, Kathiresan S. Plasma hdl cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet. 2012;380:572–80.
Riwanto M, Rohrer L, Roschitzki B, Besler C, Mocharla P, Mueller M, Perisa D, Heinrich K, Altwegg L, von Eckardstein A, Lüscher TF, Landmesser U. Altered activation of endothelial anti- and proapoptotic pathways by high-density lipoprotein from patients with coronary artery disease: role of high-density lipoprotein-proteome remodeling. Circulation. 2013;127:891–904.
Speer T, Rohrer L, Blyszczuk P, Shroff R, Kuschnerus K, Krankel N, Kania G, Zewinger S, Akhmedov A, Shi Y, Martin T, Perisa D, Winnik S, Muller MF, Sester U, Wernicke G, Jung A, Gutteck U, Eriksson U, Geisel J, Deanfield J, von Eckardstein A, Luscher TF, Fliser D, Bahlmann FH, Landmesser U. Abnormal high-density lipoprotein induces endothelial dysfunction via activation of toll-like receptor-2. Immunity. 2013;38:754–68.
Jahangiri A. High-density lipoprotein and the acute phase response. Curr Opin Endocrinol Diabet Obesity. 2010;17:156–60.
Acknowledgments
WM has received research grants, consulting fees, support for travel, and honoraria for writing this manuscript from Roche Pharma, Basel, Switzerland. MEK, TBG, UK, and GS have no potential conflicts of interest that might be relevant to the content of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kleber, M.E., Grammer, T.B., Kassner, U. et al. Dusty Punch Cards and an Eternal Enigma: High-Density Lipoproteins and Atherosclerosis. Drugs 74, 513–520 (2014). https://doi.org/10.1007/s40265-014-0189-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-014-0189-5