
Abstract
Introduction
Fluenz Tetra is an intranasal quadrivalent live attenuated influenza vaccine (QLAIV) and is recommended as the vaccine of choice for children in the 2014/2015 influenza season vaccination programme in the UK.
Objective
The primary objective of the study was to estimate the crude incidence rate of adverse events of interest (AEIs) following vaccination with the nasal QLAIV early in the 2014/2015 influenza season in children and adolescents in England.
Methods
A pilot non-interventional cohort post-authorisation safety study (PASS) was conducted during the 2014/2015 influenza season in England. Vaccinees were recruited via the mass vaccination programme in England. Participant outcomes, validated by a healthcare professional (general practitioner) where appropriate, were captured through questionnaires (surface mail, telephone, e-questionnaire). Data analysis comprised descriptive statistics and calculation of event risks and incidence rates, stratified by age group and selected co-morbidities.
Results
The final evaluable cohort consisted of 385 participants; the median (interquartile range) age was 4 (3–9) years with a range of 2–17 years, and 53.2 % were female. The most frequently reported AEI was nasal congestion (n = 167; 43.4 %; 312.3 per 1000 patient-weeks [95 % CI 267.3–364.8]). Further frequently reported AEIs were malaise (n = 87; 22.6 %; 123.4 per 1000 patient-weeks [95 % CI 98.9–154.1]) and cough (n = 80; 20.8 %; 118.5 per 1000 patient-weeks [95 % CI 95.1–147.8]). Five hypersensitivity-type reactions were reported, although on follow-up none were true hypersensitivity reactions or required hospitalisation. No serious adverse events (SAEs) were reported, with no hospitalisations or deaths. No significant change in reactogenicity or other apparent safety signals was detected as part of this study.
Conclusion
The pilot study showed no significant change in reactogenicity or other apparent safety signals from the data collected. Continued enhanced surveillance of seasonal influenza vaccines will ensure their ongoing safety for the prevention of serious illness from seasonal influenza outbreaks.
Similar content being viewed by others


Frequently reported adverse events of interest after administration of nasal quadrivalent live attenuated influenza vaccine were nasal congestion, malaise and cough. |
No serious adverse events were reported during this study. |
1 Introduction
Fluenz Tetra (Marketing Authorisation Holder (MAH): MedImmune LLC, Netherlands) is a quadrivalent live attenuated influenza vaccine (QLAIV) administered by the intranasal route and supplied in an applicator that allows a divided dose to be administered in each nostril. The device allows intranasal vaccination to be performed without the need for additional training. Neither dose needs to be repeated if the patient sneezes or blows their nose following administration [1]. The Joint Committee on Vaccination and Immunisation (JCVI) recommended that Fluenz Tetra be used as the vaccine of choice for children in the 2014/2015 flu season vaccination programme in the UK [2].
In April 2014, the European Medicines Agency (EMA) issued interim guidance on enhanced safety surveillance for seasonal influenza vaccines in the EU [3]. This guidance was issued to outline principles to be followed for improved continuous routine surveillance and to rapidly detect any increased local and systemic adverse events of interest (AEIs) or other unexpected adverse immune response that may arise during the influenza vaccine product life-cycle. In response to the guidance, a pilot active safety surveillance study was conducted as a non-interventional cohort post-authorisation safety study (PASS) for one particular influenza vaccine. The vaccine investigated, nasal QLAIV, marketed as Fluenz Tetra in the UK, is licensed for use in children and adolescents between the ages of 2 and 17 years, except in those where contraindications or special warnings and precautions for use apply. An expedited safety summary report was required as part of this guidance, with interim results available for review 1 month after first vaccination. For this reason, studies using secondary data sources would not be appropriate due to the time lag in availability of such data.
The primary objective of the study was to estimate the crude incidence rate of AEIs following vaccination with the nasal QLAIV early in the 2014/2015 influenza season in children and adolescents in England. The AEIs listed as part of the EMA guidance and pertinent to QLAIV include fever, vomiting and nausea, malaise, headache, irritability and crying (for under 5-year-old vaccinees), decreased appetite, rash, mylagia, events indicative of allergic and hypersensitivity reactions, nasal congestion, wheezing, oropharyngeal pain, cough and epistaxis. Secondary study objectives included the characterisation of patients vaccinated with nasal QLAIV, the quantification, description and classification of reported AEIs, and the characteristics of patients in whom the reported AEIs occurred.
2 Methods
2.1 Recruitment
In total, 23 general practitioner (GP) practices, 17 primary schools and six high schools across England took part in the study. Recruitment opened on 30 September 2014 and closed on 19 December 2014. Study participants were vaccinated with QLAIV between 30 September 2014 and 11 December 2014. A full study database lock was implemented on 2 January 2015 to permit receipt of responses from vaccines consented at the end of the recruitment period.
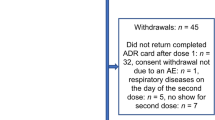
Figure 1 depicts how participants were recruited to the study through the mass vaccination programme implemented in GP practices and also through schools involved in the pilot vaccination scheme.

Flow diagram illustrating the study design and the involvement of study participants. DSRU Drug Safety Research Unit, GP general practitioner, ICF informed consent forms, PIS participant information sheets, SAEs serious adverse events
Eligible participants were children and adolescents aged 2–17 years administered with QLAIV (exposure) at participating vaccination sites. Vaccinees with co-morbidities, who take medication, or who received additional vaccines on the same day or within 1 month of receiving QLAIV were included since this was an observational cohort study conducted in a naturalistic setting. The target number of consented vaccinees was 200 vaccinees per age group, i.e. (i) 2–4 years; (ii) 5–10 years; and (iii) 11–17 years to give a target total of 600 consented vaccinees. This was to ensure that a minimum of 100 vaccinees, with completed data, per age group was reached, allowing for participants lost to follow-up and withdrawal of consent. The rationale for this sample size was based on the information provided within the interim guidance on enhanced safety surveillance [3], which requested 100 participants within each age group.
2.2 Ethical and National Health Service Approval
The Proportionate Review Sub-committee of the West of Scotland 5 Research Ethics Committee (REC) gave a favourable opinion for the conduct of the study (REC reference: 14/WS/1067) and the study was also adopted by the UK National Health Service (NHS) National Institute for Health Research (NIHR) Clinical Research Network (CRN). In addition, appropriate local NHS Research and Development permissions were granted before the study commenced.
2.3 Outcome Data
Variables assessed were derived from the responses by vaccinees or their representatives on informed consent forms, enrolment questionnaires and day 14 questionnaires (Electronic Supplementary Material 1). Participant data on demographics (age, sex), date of vaccination, relevant co-morbidities, medication, changes to medication or new medication within the 14 days post-vaccination, reported targeted AEIs (outcome), date of onset, date of resolution and other adverse events (outcome) were collected. Events were not validated by a healthcare professional unless further information was required. As shown in Fig. 1, follow-up information could be obtained either from the participant after receipt of their day 14 data or with their GP using a standard follow-up questionnaire or a telephone call.
All information on the e-questionnaires completed on the bespoke study website (via web-portal) was automatically transferred to a Drug Safety Research Unit (DSRU) database. Information completed on paper questionnaires was entered onto the DSRU database by suitably trained data entry/coding DSRU staff. Events collected as free-text were coded onto the database using the MedDRA® dictionary. Analyses were performed using STATA® version 12 (StataCorp, College Station, TX, USA).
2.4 Data Analysis
Data were summarised using univariate descriptive statistics, including measures of central tendency and dispersion for continuous variables (mean, standard deviation [SD], median, range and percentiles) and frequencies with proportions for categorical variables. The continuous variable of age (at vaccination) was categorised into age groups (2–4, 5–10 and 11–17 years). Adverse event of interest incidence risks were calculated with 95 % exact binomial confidence intervals (CIs). Person-time at risk for each event was calculated, along with incidence rates per 1000 patient-weeks and the 95 % exact Poisson CI. Stratification of AEI risks and rates was performed by age group and by medical condition, where appropriate. Since participants were asked to provide information on new symptoms, events were assumed to be incident reports post-vaccination.
An assessment of missing information for all key variables on paper questionnaires was conducted during the data analysis stage to determine the distribution of missingness and whether there was pattern of missingness that showed any dependency on age group. A sensitivity analysis was planned a priori to examine the impact of missing data on estimates of risk and incidence rates if proportion of missingness was greater than 20 %. Since this was a pilot study, and given the small sample size, it was decided that methods to handle missing data would not be appropriate unless there was a very large amount of missing data. Questionnaires completed online did not have missing data as the participant was prompted to complete each field before moving onto the next.
3 Results
3.1 Cohort Characteristics
Participant recruitment is summarised in Fig. 2. The final evaluable cohort comprised 385 participants (see Table 1 for characteristics of the evaluable cohort).

Flow of participant recruitment. ICF informed consent forms
An assessment of the level of missing data was carried out on the evaluable cohort. No more than 5 % of data were missing for any main question or sub-question on the questionnaires (data not presented). Therefore, no sensitivity analysis was conducted.
The median age of the evaluable cohort was 4 years, with an interquartile range (IQR) of 3–9 years. The median age for females was 5 years (IQR 3–9 years) and for males was 4 years (IQR 3–9 years). There were 205 (53.2 % of cohort) females and 180 (46.8 % of cohort) males in the evaluable cohort.
Five vaccinees had additional vaccinations on the same day as receiving QLAIV (Fluenz Tetra) and a further five vaccinees reported receiving an additional vaccine or additional vaccines within 1 month of receiving QLAIV (Fluenz Tetra). These vaccinations were the MMR (mumps, measles and rubella), 4-in-1 Preschool Booster (DTaP/IPV [diphtheria, tetanus, pertussis, inactivated polio) and HPV (human papilloma virus) vaccine.
Of the total evaluable cohort, 85 participants reported that the vaccinee had pre-existing asthma, with only two participants having pre-existing diabetes mellitus, two with pre-existing heart disease, one with pre-existing chronic kidney disease and one with pre-existing immunosuppression. Participants reported that 92 (23.9 %) of the evaluable cohort take prescribed or over-the-counter medications, the majority of which were to treat respiratory conditions: 66 of 92 receiving salbutamol (Anatomical Therapeutic Chemical [ATC] Drug Class R03AC) and 42 of 92 receiving beclometasone dipropionate (ATC Drug Class R03BA]). This correlates to the high proportion of the evaluable cohort who reported having pre-existing asthma. Five participants reported a change to their medication in the 14 days post-vaccination.
The study was designed to capture the events experienced by children and adolescents following their first vaccination with QLAIV (Fluenz Tetra). When questioned, GP practices indicated that only a very small proportion of vaccinees or no vaccinees received two doses of QLAIV (Fluenz Tetra). The highest proportion of those vaccinated receiving two doses at any one GP practice was 3.5 % (14/400) of the total vaccinated with QLAIV (Fluenz Tetra), with the majority of practices indicating that only two or three children received two doses. No participants indicated on their questionnaire in the free-text event section or anywhere on the paper version that they received a second dose of QLAIV (Fluenz Tetra) or that they experienced any events following a second dose of QLAIV (Fluenz Tetra).
3.2 Adverse Events of Interest
The total number of AEIs reported in the evaluable cohort was 650 in 237 participants. Targeted AEIs are summarised in Table 2 and other events are summarised in Table 3. The most frequent AEI across the whole evaluable cohort was nasal congestion (n = 167; 43.4 %). Furthermore, more than 20 % of the cohort reported AEIs of malaise (n = 87; 22.6 %) and cough (n = 80; 20.8 %). In contrast, less than 5 % of the cohort reported AEIs of wheezing (n = 9; 2.3 %), nosebleed (n = 9; 2.3 %), rash (n = 8; 2.1 %) and hypersensitivity-type reactions (n = 5; 1.3 %).
Four of the five vaccinees who received additional vaccines on the same day as QLAIV reported experiencing AEIs within the observation period (high temperature n = 3, malaise n = 3, decreased appetite n = 3, muscle/joint pain n = 1, wheezing n = 4, nasal congestion n = 4, mouth/throat pain n = 3, cough n = 1, increased irritability n = 4, increased crying n = 2).
In total, five counts of hypersensitivity-type reactions were recorded in five separate patients, but on follow-up none were true hypersensitivity reactions. Follow-up information indicated that four of the five participants described “feeling dizzy” or “light headed” post-vaccination which quickly resolved, hence their reason for ticking yes to the question “Did the vaccinee experience any of the following: swelling of the face, lips or tongue, difficulty breathing, feeling of dizziness/light-headedness, general itchiness with a rash”.
Crude incidence risks and rates for AEIs were calculated for each age group and are presented in Table 4. Total time at risk is the denominator of person-time at risk censored at the time of the AEI.
Events with crude incidences highest in the 2–4 years age group compared with other age groups were nasal congestion (346.4 per 1000 person-weeks [95 % CI 280.1–428.5]) and decreased appetite (91.2 per 1000 person-weeks [95 % CI 63.8–130.5]). Crude incidence rates highest in the 11–17 years age group compared with other age groups were nausea and vomiting (40.4 per 1000 person-weeks [95 % CI 18.2–90.0]) and headache (127.9 per 1000 person-weeks [95 % CI 78.3–208.7]). In addition to nausea and vomiting and headache, crude incidence rates of generally feeling unwell (malaise), mouth and throat pain and wheezing increased with age. AEIs for which the rate consistently decreased with age included high temperature and cough.
In addition to the targeted event reports, a total of 35 “other” non-targeted events were reported in 26 participants. Seven counts of tiredness (preferred term [PT] fatigue) were reported in the evaluable cohort and seven events were reported as classified within the gastrointestinal disorders System Organ Class (SOC) (PTs: diarrhoea, mouth ulceration, abdominal pain upper, anal inflammation, chapped lips, dysphagia). One participant reported experiencing flu symptoms post-vaccination. One participant experienced a rash that was described as “blister like spots” and coded as a MedDRA® PT of “blister” on the day of vaccination.
There were no reported pregnancies during the course of the study. There were also no deaths reported during the course of the study.
In terms of serious adverse events (SAEs) or important medical events, none were recorded during the course of the study. Additionally, no events reported are listed as potential risks in the Risk Management Plan (RMP).
Stratification of incidence risks and rates of AEIs by medical condition was performed (data not presented). Results were stratified for participants with and without asthma (Electronic Supplementary Material 2, Supplementary Table 1) as 22 % (n = 85) of the evaluable cohort indicated they had the condition. Incidence rates of wheezing were approximately 30 times higher in the asthma group (50.1 per 1000 patient-weeks [95 % CI 25.1–100.2]) than in participants without asthma (1.7 per 1000 patient-weeks [95 % CI 0.2–11.9]).
Furthermore, upon examination of batch effects on the counts of AEIs, no definitive pattern or clustering of AEIs per batch in the 14 different batches recorded for vaccinees with AEIs was visible (data not presented).
4 Discussion
This pilot study has enabled the collection of important safety data from participants vaccinated with QLAIV (Fluenz Tetra) within GP practices and schools in pilot areas across England. The study fulfils the regulatory objectives for enhanced safety surveillance for seasonal influenza vaccines in the early detection of selected events of interest.
The most frequently reported AEI across all age groups within this study was nasal congestion, a very common adverse effect (frequency ≥1 in 10) according to the Summary of Product Characteristics (SmPC) [1]. Other frequently reported AEIs were malaise and decreased appetite, which are also specified as very common in the SmPC [1]. Cough, a common AEI within this study, on the other hand, does not feature in the SmPC. However, in a randomised double-blind non-inferiority study of QLAIV in 1200 subjects aged 18–49 years, cough was reported by 13.6 % of the safety cohort within 14 days post-vaccination with nasal QLAIV [4]. Within this same study, runny/stuffy nose (43.6 %) was the most frequently reported symptom within the safety cohort 14 days post-vaccination [4]. In a further study of QLAIV in the USA using the Vaccine Adverse Event Reporting System (VAERS), 8 % of all 599 spontaneous reports in 2- to 17-year-olds were reports of cough [5]. Studies of the trivalent LAIV demonstrated similar patterns of AEIs, with nasal congestion (also referred to as runny nose or rhinorrhoea) being the most frequently reported AEI among children [6, 7] as well as adolescents and adults [8].
All five reported events of hypersensitivity-type reactions within this study were not deemed to be true hypersensitivity reactions, with vaccinees reporting symptoms of “dizziness” or “light-headedness” upon follow-up. The wording of the question on the day 14 questionnaire may have been misleading as it did not explicitly ask for symptoms corresponding to an allergic response, but any of the following: “Swelling of the face, lips or tongue, difficulty breathing, feeling of dizziness/light-headedness, general itchiness with a rash”. Hypersensitivity-type reactions have been rarely reported with live attenuated influenza vaccines (LAIV). A single event of bronchospasm was reported following vaccination with the tetravalent LAIV (TLAIV) [4]. Furthermore, a single report of “allergy” within VAERS in 2- to 17-year-olds vaccinated with QLAIV was identified, which was a non-anaphylaxis allergic reaction, as well as a single report of anaphylaxis within 18- to 49-year-olds [5]. Five reports of “allergy” within VAERS in 2- to 17-year-olds vaccinated with trivalent LAIV included three reports of anaphylaxis and two non-anaphylaxis allergic reactions [5], and five reports in 18- to 49-year-olds included two reports of anaphylaxis and three non-anaphylaxis allergic reactions [9].
No SAEs or important medical events were reported during the course of the study and no events were considered to be important identified or potential risks. Spontaneous reports within VAERS showed similar results, with no concerning safety patterns relating to QLAIV vaccination [5].
A large number of study participants reported having asthma, which is a special warning/precaution for use within the SmPC, which states that QLAIV (Fluenz Tetra) should not be administered to children or adolescents with severe asthma or active wheezing because these individuals have not been adequately studied in clinical studies [1]. Severe asthma or active wheezing was not reported as a free-text event as being present at the time of vaccination or following vaccination. The high incidence rates of wheezing within the asthma group following vaccination are potentially a result of the aetiology of disease and not the vaccination. The use of the vaccine within asthmatic children has been reported in other studies [5].
This active surveillance study enabled the successful monitoring of safety of seasonal influenza vaccines. Questionnaires were completed well online, and there were minimal missing data on paper questionnaires. Similar methodologies have also been utilised successfully in other countries. In New Zealand, telephone surveys were used to successfully evaluate febrile events following vaccination of individuals from 184 GP surgeries with five different influenza vaccines [10]. An Australian study used web-based surveys to explore a number of AEIs following vaccination of individuals from 15 GP surgeries with five different influenza vaccines [11]. Response rates for this web-based survey (61 %) were similar to the response rates within the online questionnaire group within our study (66.2 %) [11]. The study enabled information on AEIs to be captured directly from vaccinees, hence capturing AEIs that are not medically attended or reported to the GP.
The cohort involved in this study is thought to be representative of the population vaccinated with QLAIV (Fluenz Tetra) during the 2014/2015 influenza season. Participants were recruited from a good geographical spread of GP practices across England as well as from primary and high schools in selected pilot areas. The cohort included both female and male children and adolescents with co-morbidities in the age range 2–17 years. Open inclusion criteria within this study allowed the investigators to gain an understanding of the safety of the vaccine in a naturalistic setting.
This study is a pilot and will enable, in conjunction with other studies, the refinement of the thinking and methodologies of seasonal influenza vaccine safety surveillance. In future work, continued safety surveillance of seasonal influenza vaccines remains a priority as a result of continued antigenic drift and shift of the influenza virus [12].
4.1 Limitations
There are some limitations to this study. First, fewer children aged 11–17 years were recruited than other age groups. This is probably due to Public Health England’s recommended usage of the vaccine [2]. During the 2014/2015 flu season, only “high risk” individuals, i.e. those at risk of serious illness or death, as defined by the Department of Health seasonal influenza immunisation programme specifications [13], and those in pilot area schools were eligible for vaccination in England. Concerns from teaching staff and immunisation coordinators about low vaccine uptake by children and adolescents in schools may have had a negative impact on their eagerness to participate in this pilot study. This demographic distribution pattern may change in subsequent years as the vaccine is rolled out to more healthy children [2]. Additionally, the majority of GPs involved in this study vaccinated mostly children under 6 years old with QLAIV (Fluenz Tetra) and very few between 6 and 17 years of age. It is unknown whether adolescents received different influenza vaccinations at these practices. Only a small number of GPs, primary and high schools were recruited into the study, which is a result of the desired sample size for the study based on interim guidance from the EMA [3].
In addition, the number of participants from the same family was not recorded, and so it is unclear if there may be clustering of AEIs within families.
Another limitation is that the study focused on the detection of common AEIs that manifest in the short-term post-vaccination, which is indicative of increased reactogenicity, as laid out in the interim guidance, rather than uncommon reactions. Other study designs may be more appropriate for the detection of rare adverse events. For example, case-control studies have been used to identify uncommon adverse events such as Bell’s palsy following vaccination with inactivated influenza vaccines [14]. Furthermore, the study was not designed to assess causality between the vaccine and adverse events.
Response rates ranged from 66 to 86 % in this study, resulting in the potential for non-response bias. However, the response rate in this study is higher than the reporting rates of suspected adverse drug reactions in the Yellow Card Scheme [15].
Finally, the study questionnaires only captured safety events for those receiving their first dose of QLAIV (Fluenz Tetra). It is, however, thought that only a very small proportion of vaccinees would have received two doses. The SmPC advises that a second dose should be given after an interval of at least 4 weeks in children who have not previously been vaccinated against seasonal influenza [1]; however, the Public Health England Flu Plan specifies that only children in clinical risk groups aged 2 to <9 years who have not received an influenza vaccine before should be offered a second dose of the nasal QLAIV Fluenz Tetra (given at least 4 weeks apart), which may account for low numbers receiving a second dose [2].
5 Conclusion
No significant change in reactogenicity or other apparent safety signal from the data collected has been detected as part of this study. This study has shown that data can be generated quickly by gathering vaccinee- or participant-level feedback following vaccination using both an online survey and through the use of a well-completed simple paper questionnaire. Follow-up with participants or their GP is also possible using this methodology. Continued enhanced surveillance of seasonal influenza vaccines will ensure their ongoing safety for the prevention of serious illness from seasonal influenza outbreaks.
References
Limited AstraZeneca UK. Summary of product characteristics: Fluenz Tetra nasal spray suspension Influenza vaccine (live attenuated, nasal). Luton: AstraZeneca; 2015.
Department of Health, Public Health England, NHS England. Flu plan: winter 2014/15. 2014. https://www.gov.uk/government/uploads/system/uploads/attachment_data/file/306638/FluPlan2014_accessible.pdf. Accessed 18 Dec 2015.
European Medicines Agency. Interim guidance on enhanced safety surveillance for seasonal influenza vaccines in the EU. EMA/PRAC/222346/2014. 2014. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/04/WC500165492.pdf. Accessed 18 Dec 2015.
Block SL, Yi T, Sheldon E, Dubovsky F, Falloon J. A randomized, double-blind noninferiority study of quadrivalent live attenuated influenza vaccine in adults. Vaccine. 2011;29(50):9391–7.
Haber P, Moro PL, Cano M, Lewis P, Stewart B, Shimabukuro TT. Post-licensure surveillance of quadrivalent live attenuated influenza vaccine United States, vaccine adverse event reporting system (VAERS), July 2013–June 2014. Vaccine. 2015;33(16):1987–92.
Belshe RB, Mendelman PM, Treanor J, King J, Gruber WC, Piedra P, et al. The efficacy of live attenuated, cold-adapted, trivalent, intranasal influenzavirus vaccine in children. N Engl J Med. 1998;338(20):1405–12.
Bracco Neto H, Farhat CK, Tregnaghi MW, Madhi SA, Razmpour A, Palladino G, et al. Efficacy and safety of 1 and 2 doses of live attenuated influenza vaccine in vaccine-naive children. Pediatr Infect Dis J. 2009;28(5):365–71.
Ambrose CS, Wu X. The safety and effectiveness of self-administration of intranasal live attenuated influenza vaccine in adults. Vaccine. 2013;31:857–60.
Haber P, Moro PL, McNeil MM, Lewis P, Woo EJ, Hughes H, et al. Post-licensure surveillance of trivalent live attenuated influenza vaccine in adults, United States, vaccine adverse event reporting system (VAERS), July 2005–June 2013. Vaccine. 2014;32:6499–504.
Petousis-Harris H, Poole T, Turner N, Reynolds G. Febrile events including convulsions following the administration of four brands of 2010 and 2011 inactivated seasonal influenza vaccine in NZ infants and children: the importance of routine active safety surveillance. Vaccine. 2012;30(33):4945–52.
Cashman P, Moberley S, Dalton C, Stephenson J, Elvidge E, Butler M, et al. Vaxtracker: active on-line surveillance for adverse events following inactivated influenza vaccine in children. Vaccine. 2014;32(42):5503–8.
Fauci AS. Seasonal and pandemic influenza preparedness: science and countermeasures. J Infect Dis. 2006;194(Suppl 2):S73–6.
PHE. Public health functions to be exercised by NHS England: seasonal influenza immunisation programme. 2013. https://www.gov.uk/government/uploads/system/uploads/attachment_data/file/192974/13_Seasonal_flu__service_specification_VARIATION__130422.pdf
Martin RM, Kapoor KV, Wilton LV, Mann RD. Underreporting of suspected adverse drug reactions to newly marketed (“black triangle”) drugs in general practice: observational study. BMJ. 1998;317(7151):119–20.
Mutsch M, Zhou W, Rhodes P, Bopp M, Chen RT, Linder T, et al. Use of the inactivated intranasal influenza vaccine and the risk of Bell’s palsy in Switzerland. N Engl J Med. 2004;350(9):896–903.
Acknowledgments
The authors acknowledge the support of the National Institute of Health Research Clinical Research Network (NIHR CRN). The Drug Safety Research Unit (DSRU) also wishes to acknowledge the contribution of Bob Brody (AstraZeneca) and Fei Ji (AstraZeneca) for their assistance with the specification of the Statistical Analysis Plan, provision of data from the clinical programme that was used for comparison purposes and their advice and commentary on the final results.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The study was funded by AstraZeneca. The DSRU is an independent charity (No. 327206), which works in association with the University of Portsmouth (Portsmouth, UK). It receives unconditional donations from pharmaceutical companies. The companies have no control on the conduct or the publication of the studies conducted by the DSRU. The DSRU has received such funds from the manufacturer of Fluenz Tetra (AstraZeneca).
Conflict of interest
Rhian McNaughton, Elizabeth Lynn, Vicki Osborne, Abigail Coughtrie and Deborah Layton have no conflicts of interest directly relevant to the content of this study. Saad Shakir reports personal fees from ICON (contract research organization), Shire Pharmaceuticals, ONO Pharmaceuticals, Intermune Pharma and IPSEN, outside the submitted work.
Ethical approval
The study was approved by the Proportionate Review Sub-committee of the West of Scotland 5 Research Ethics Committee (REC) (REC reference: 14/WS/1067).
Patient consent
Patients, or their parent or legal guardian for children under 16 years of age, were asked to complete a consent form prior to participation in the study. Only patients with a completed consent form returned to the DSRU were included in the study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
McNaughton, R., Lynn, E., Osborne, V. et al. Safety of Intranasal Quadrivalent Live Attenuated Influenza Vaccine (QLAIV) in Children and Adolescents: A Pilot Prospective Cohort Study in England. Drug Saf 39, 323–333 (2016). https://doi.org/10.1007/s40264-015-0384-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40264-015-0384-7