
Abstract
Background
Patients with multiple sclerosis (MS) experience relapses and sustained disability progression. Since 2004, the number of disease-modifying therapies (DMTs) for MS has grown substantially. As a result, patients, healthcare providers, and insurers are increasingly interested in comparative efficacy and safety evaluations to distinguish between treatment options, but head-to-head studies between DMTs are limited.
Objective
The aim of the current study was to compare efficacy and safety outcomes with the DMTs ozanimod and dimethyl fumarate (DMF) using a matching-adjusted indirect comparison (MAIC) to adjust for cross-trial differences in study design and population.
Methods
A systematic literature review was performed to identify clinical studies evaluating the efficacy and safety of ozanimod compared with DMF. Individual patient-level data (IPD) for ozanimod were obtained from the SUNBEAM and RADIANCE Part B trials, and aggregate-level patient data (APD) for DMF were obtained from CONFIRM and DEFINE. A MAIC is used to weight IPD to APD based on important baseline patient characteristics considered to be effect modifiers or prognostic factors in order to balance the covariate distribution to establish more homogenous trial populations. Once trial populations are determined to be sufficiently homogenous, outcomes of interest are estimated and used to generate treatment effects between the weighted IPD and APD. We used MAIC methodology to compare efficacy and safety outcomes of interest between ozanimod 1.0 mg once daily (OD) and DMF 240 mg twice daily (BID), including confirmed disability progression (CDP) at 3 and 6 months, annualized relapse rate (ARR), proportion of patients relapsed, overall adverse events (AEs), serious AEs (SAEs), and discontinuations due to AEs.
Results
After matching patient data, baseline patient characteristics were balanced between patients receiving ozanimod and those receiving DMF. Compared with DMF, ozanimod demonstrated significantly improved CDP at 3 months (hazard ratio 0.67; 95% confidence interval [CI] 0.53–0.86), ARR (rate ratio [RR] 0.80; 95% CI 0.67–0.97), proportion of patients relapsed (odds ratio [OR] 0.66; 95% CI 0.52–0.83), overall AEs (OR 0.11; 95% CI 0.08–0.16), SAEs (OR 0.27; 95% CI 0.19–0.39), and discontinuations (OR 0.11; 95% CI 0.07–0.17). CDP at 6 months did not differ significantly between the two agents (RR 0.89; 95% CI 0.62–1.26).
Conclusions
After adjustment of baseline patient characteristics, the MAIC demonstrated that the efficacy and safety of ozanimod 1.0 mg OD was superior to that of DMF 240 mg BID. Although a MAIC is less likely to produce biased estimates than a naïve or a standard indirect treatment comparison via a common comparator, limitations include potential confounding due to unobserved and thus unaccounted for baseline differences.
Plain Language Summary
Ozanimod and dimethyl fumarate (DMF) are disease-modifying therapies used to treat relapsing-remitting multiple sclerosis (MS). Comparative efficacy and safety evaluation is important to key patients, healthcare providers, and health insurers; however, head-to-head studies between MS therapies are limited. In this analysis, we used an indirect treatment comparison method, specifically a matching-adjusted indirect comparison (MAIC), to compare results of clinical trials of ozanimod and DMF. In this MAIC, findings suggested that ozanimod was associated with greater reductions of relapses, a lowered risk of disability progression at 3 months, and improved safety outcomes compared with DMF. Although MAICs were conducted while adjusting for important treatment-effect modifiers and/or prognostic factors, the possibility of confounding as a result of unobserved baseline differences remains. Such an issue can be resolved only by conducting a head-to-head treatment comparison in a randomized clinical trial.
Similar content being viewed by others

Digital Features To view digital features for this article, go to https://doi.org/10.6084/m9.figshare.14182724. |
In the absence of head-to-head clinical trials, indirect treatment comparison methods provide additional evidence to inform decision making. |
After adjustment of baseline patient characteristics, ozanimod demonstrated improved relapse outcomes, lower risks of adverse outcomes, and lower discontinuation rates compared with dimethyl fumarate. Ozanimod demonstrated significantly improved confirmed disability progression (CDP) at 3 months. There were no significant differences in CDP at 6 months. |
1 Introduction
Multiple sclerosis (MS) is a chronic inflammatory, neurodegenerative, central nervous system (CNS) disease characterized by demyelination and axonal and neuronal loss. MS most commonly presents as a relapsing-remitting MS (RRMS) phenotype [1,2,3,4]. Although patients with relapsing MS (RMS) may experience recovery after acute episodes, worsening residual disability may accumulate over time [3,4,5,6]. The number of immunomodulating disease-modifying therapies (DMTs) available for RMS is growing [5, 7]. These immunomodulators may reduce the frequency and severity of relapses, reduce evidence of continued disease activity on magnetic resonance imaging, and, in some instances, significantly reduce the accumulation of sustained increases in disability [2, 7,8,9].
Dimethyl fumarate (DMF; Tecfidera, Biogen Inc., Cambridge, MA, USA) exerts anti-inflammatory and cytoprotective effects arising, at least in part, from the activation of the nuclear 1 factor (erythroid-derived 2)-like 2 (Nrf2) antioxidant response pathway; DMF may also play a part in modulating immune cell responses [10, 11]. Clinical studies have demonstrated that DMF treatment results in reduced absolute lymphocyte counts (ALCs) [12]; however, the reductions are not uniform, with the greatest decrease seen in T-cell lymphocytes and, to a lesser extent, in B cells and natural killer (NK) cells [13,14,15]. DMF reduces circulating T cells but also shifts cell polarity from proinflammatory T-helper type 1 (Th1) and Th17 phenotypes toward anti-inflammatory Th2 cells [16] and shifts B cells toward tolerogenic phenotypes [17]. Within 8 weeks of starting DMF in patients with MS, there was not only a drop in cluster of differentiation (CD)-4+ and CD8+ T cells and non-class switched B cells but also an increase in immunoregulatory CD56+bright NK cells [18]. No effect was seen on serum immune globulin levels [18].
DMF is believed to increase the bioavailability of the antioxidant glutathione (GSH), to react with thiols on cysteine-containing neuronal and astrocytic proteins [19, 20], and to modulate microglial activation [21]; however, these latter actions would require substantial entrance into the CNS by the DMF active metabolite, monomethyl fumarate, but little is known about the actions of this metabolite within the CNS in vivo at present.
It is well-known that DMF treatment mobilizes the transcription factor Nrf2 oxidative stress response pathway, which maintains redox homeostasis and reduces reactive oxygen species (ROS) [20, 22], to lower oxidative stress [23, 24]. Independent of its effects on the Nrf2 and nuclear factor kappa B B-cell activation pathways, DMF induces anti-inflammatory signaling in macrophages and microglia [25, 26]. In contrast to earlier work, more recent studies have demonstrated that oxidative stress biomarkers in blood are increased by DMF, with parallel upregulation of oxidative stress genes [27]. A recent analysis showed DMF treatment was associated with increased monocyte counts, and therapeutic clinical response in patients with MS treated with DMF was associated with both elevated monocyte counts and reduced ALCs [18]. Furthermore, DMF increased the expression of ROS genes and ROS production by monocytes, which was in turn associated with disease stabilization in patients with MS [28]. DMF also suppresses the Th17 regulator transcription factor RORϫt and promotes a shift toward cytotoxic T-cell gene expression. Anti-CD8+ Th17 cell activity was dependent on ROS expression and was blocked by GSH. Thus, CD8+Th17 cell responsiveness to DMF appears to be ROS dependent [28].
Several clinical trials (CONFIRM, DEFINE, and APEX) have demonstrated the clinical safety and efficacy of DMF, which was approved by the US FDA in 2013 and the European Medicines Agency in 2014 as a treatment for RRMS [10, 11, 29,30,31]. In DEFINE, the annualized relapse rate (ARR) at 2 years showed significant relative reductions with DMF of 53 and 48% when given twice daily (BID) and three times daily (TID), respectively, compared with placebo [11]. DMF significantly reduced the risk of sustained disability worsening (confirmed disability progression [CDP]) at 2 years by 38% in the DMF BID group and by 34% in the DMF TID group [11]. In CONFIRM, the ARR at 2 years was significantly reduced with DMF BID and TID regimens (44 and 51%, respectively) compared with placebo [10]. In CONFIRM, sustained worsening of disability status was not significantly reduced with DMF treatment compared with placebo [10]. The most commonly reported adverse events (AEs; incidence ≥ 10% and ≥ 2% vs. placebo) in clinical trials were flushing, abdominal pain, diarrhea, and nausea [30].
Ozanimod (Zeposia, Bristol Myers Squibb, Princeton, NJ, USA) is a sphingosine 1-phosphate (S1P) receptor modulator that binds with high affinity selectively to S1P receptors 1 and 5 (S1PR1, S1PR5) [32]. Inhibition of S1PR1 impairs B- and T-cell lymphocyte egress from peripheral lymphoid organs [32,33,34]. As an inhibitor of S1PR1, ozanimod also has the potential to downregulate astrocyte activation, inhibit proinflammatory activation of myeloid cells by inhibiting their expression of major histocompatibility complex-II and CD40, and reduce antigen presentation. S1PR1 antagonism also impairs interleukin-6-driven Th17 and Th1 lymphocyte polarity [35,36,37,38,39]. S1PR5 modulation has been shown to increase blood–brain barrier integrity by tightening CNS endothelial cell junctions [40]. S1PR1 modulation may also promote oligodendroglial progenitor cell migration and maturation and promote survival and myelin production by oligodendroglia [41, 42]. Ozanimod was approved in 2020 for the treatment of RRMS [43, 44]. Data from the phase III RADIANCE Part B (RADIANCE-B) [45] and SUNBEAM [46] clinical trials demonstrated the efficacy and safety of ozanimod in patients with RMS. The ARR at study end was significantly lower for patients treated with DMF than for those receiving interferon (IFN)-β-1a in both trials (0.17 vs. 0.28 [P < 0.0001] in RADIANCE-B and 0.18 vs. 0.35 [P < 0.0001] in SUNBEAM) [45, 46]. In the pooled analysis of RADIANCE and SUNBEAM, the proportions of participants with CDP at 3 months and at 6 months were not significantly different between treatment groups [45]. The most commonly reported AEs included upper respiratory infection, hepatic transaminase elevation, orthostatic hypotension, urinary tract infection, back pain, hypertension, and upper abdominal pain [44].
Patients, healthcare providers, and health insurers would be assisted by comparative efficacy and safety evaluations to distinguish between the different treatment options for RMS, but head-to-head studies between DMTs are limited. Indirect treatment comparisons are generally used to compare treatments for which head-to-head data are not available; however, such network meta-analyses (NMAs) have limitations. One of the key assumptions of an NMA is that treatment-effect modifiers are equally distributed across trials. However, in the ozanimod and DMF pivotal trials, observed differences in baseline patient characteristics between populations were considered important treatment-effect modifiers and prognostic factors in MS, underscoring a key limitation of results derived from NMA methodology. An alternative strategy is to employ matching-adjusted indirect comparison (MAIC), which can be used when no existing head-to-head trials have compared treatments of interest and there is a disconnected network (due to a lack of a common comparator or single-arm studies) or there are observed imbalances in baseline patient characteristics thought to be treatment-effect modifiers between trials [47].
Recent analyses have used MAIC methodology to adjust for potential cross-trial differences in patient populations to compare treatments for MS [48,49,50]. In the absence of head-to-head clinical trials, MAIC analyses have been conducted comparing fingolimod versus DMF, cladribine versus alemtuzumab, and ozanimod versus fingolimod [48,49,50]. Because no studies have directly compared ozanimod and DMF in the treatment of patients with RMS, the aim of the current study was to assess efficacy and safety outcomes of interest between ozanimod 1.0 mg daily (OD) and DMF 240 mg BID using MAIC methodology.
2 Methods
2.1 Data Source
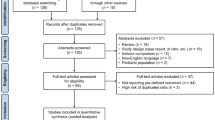
A systematic literature review was performed to identify clinical studies evaluating the efficacy and safety of ozanimod versus DMF. The review, conducted in June 2019, identified and extracted aggregate-level patient data (APD) from DMF trials of at least 1 year in duration. Exclusion criteria for individual studies included those lacking outcomes data for the primary endpoints of interest and studies that diagnosed MS using the Poser criteria, which did not include the results of magnetic resonance imaging [5]. Following this process, individual-level patient data (IPD) for ozanimod were obtained from two trials: RADIANCE-B (NCT02047734 [RPC01-201B]) [45] and SUNBEAM (NCT02294058 [RPC01-301]) [46]. RADIANCE-B was a randomized, IFN-β-1a–controlled phase III trial of ozanimod in patients with RMS (N = 1313) who were randomized (1:1:1) to oral ozanimod 0.5 or 1.0 mg OD or to intramuscular IFN-β-1a 30 µg weekly for 2 years to assess ARR. SUNBEAM also evaluated ARR in an active-controlled phase III trial in 1346 patients with RMS randomized (1:1:1) to oral ozanimod 0.5 or 1.0 mg OD or intramuscular IFN-β-1a 30 µg weekly over a minimum 12-month period. In both trials, a dose-escalation regimen for ozanimod was implemented for all patients (0.25 mg on days 1–4; 0.5 mg on days 5–7; then the assigned dose of 0.5 or 1.0 mg from day 8 through week 24) to mitigate potential first-dose cardiac rate effects [50].
APD for DMF were available from two randomized, double-blind, placebo-controlled phase II clinical trials: CONFIRM (NCT00451451) and DEFINE (NCT00420212) [10, 11]. Both studies were conducted over a period of 96 weeks in 28 countries. In CONFIRM, patients with RRMS were randomized (1:1:1) to placebo, DMF 240 mg BID, or glatiramer acetate 20 mg OD, and the primary endpoint was ARR. The DEFINE study evaluated the proportion of relapsed patients among those randomized (1:1:1) to placebo or DMF 240 mg BID or TID. The APEX study was considered but not included because it had a duration of less than 1 year (24 weeks), which may have biased the results as it did not allow sufficient study duration to accrue outcomes of interest [29].
2.2 Study Outcomes
The outcomes of interest assessed in the MAIC were based on the primary and secondary endpoints of the RADIANCE-B and SUNBEAM trials and included CDP at 3 and 6 months, ARR, proportion of patients relapsed, serious AEs (SAEs), overall AEs, and discontinuations due to AEs. All adjustments were made to include only studies reporting the outcome of interest for each comparison.
2.3 Assessment of Cross-Trial Similarities and Differences
A feasibility assessment compared the trial characteristics and eligibility criteria of the ozanimod and DMF trials and evaluated patient baseline characteristics for imbalances. The ozanimod and DMF trials were randomized, double-blind, multicountry phase III studies that required patients to be aged 18–55 years with an Expanded Disability Status Scale (EDSS) score of 0.0–5.0. Additionally, trials assessing ozanimod and DMF included patients with or without prior exposure to DMTs and required discontinuation of treatment with IFN-β and/or glatiramer acetate prior to randomization.
Several cross-trial differences were observed. Both masking and comparator arms differed between the trials. The DMF trials were placebo controlled, whereas the ozanimod trials used IFN-β-1a as the comparator. Variability was also observed between the ozanimod and DMF trials across MS criteria, MS subtype, and prior treatment. Ozanimod trials required an MS diagnosis based on 2010 McDonald criteria, whereas DMF trials required an MS diagnosis based on 2005 McDonald criteria. Ozanimod trials enrolled patients with secondary progressive MS (< 0.4%) and progressive-relapsing MS (< 1.8%) in addition to patients with RRMS (> 98%), whereas DMF trials enrolled only those with RRMS. DMF trials had higher proportions of patients who had received a prior DMT (39 vs. 29%). Ozanimod trials required patients to have had either at least one relapse in the 12 months before screening or at least one relapse in the 24 months before screening with at least one gadolinium-enhancing (GdE+) lesion in the 12 months before randomization. DMF trials required patients to have had either at least one clinically documented relapse within 12 months prior to randomization or at least one GdE+ lesion within 6 weeks prior to randomization. Baseline patient characteristics also varied for sex (female: 65% ozanimod vs. 72% DMF) and disease duration from diagnosis (5–6 years ozanimod vs. 3–4 years DMF).
2.4 Statistical Methods
MAIC methodology was used to adjust for both potential treatment-effect modifiers and prognostic factors, since the trials lacked a common comparator [47, 51]. The DMF 240 mg treatment group was pooled across the CONFIRM and DEFINE studies, whereas the ozanimod 1.0 mg group was pooled across the RADIANCE-B and SUNBEAM studies. IPD from the ozanimod trials were weighted to balance covariate distribution with APD from the DMF trials. A logistic propensity score model was developed that included all relevant potential effect modifiers and prognostic factors and used the method of moments to estimate the weights for the ozanimod trials versus the DMF trials [52]. Based on clinical input and previously conducted MAICs in the RRMS population [48, 53], the current MAIC used IPD weighted by the following relevant treatment-effect modifiers: EDSS score (mean), prior relapse at 1 year (mean), GdE+ lesions (mean), prior DMTs (percentage), age (mean), and sex (proportion female). The model also included one prognostic factor, weight (mean, kilograms).
Baseline patient characteristics and selected outcomes were described and compared for ozanimod versus DMF. Baseline patient characteristics (i.e., demographic and clinical characteristics) were described using means and standard deviations for continuous variables; frequencies and percentages were reported for categorical variables. Differences in outcomes for each comparison were presented as rate ratios (RRs) for rate outcomes (e.g., ARRs) and as odds ratios (ORs) and 95% confidence intervals (CIs) for binary comparison ratios (e.g., proportion relapsed, SAEs, overall AEs, discontinuations).
CDP outcomes were assessed using hazard ratios (HRs) and RRs. The first approach was to estimate HRs using digitized Kaplan–Meier curves and the Guyot algorithm to generate pseudo-IPD data from APD trials [54, 55]. Cox proportional hazards (PHs) were used to obtain HRs using the following four steps:
-
(1)
Digitizing Kaplan–Meier curves for APD trials to generate pseudo-IPD via the Guyot algorithm [55, 56].
-
(2)
Appending pseudo-IPD to individual weighted IPD trials, as if it were one single trial. Pseudo-IPD were assigned a weight of 1, which is equivalent to “no weight.”
-
(3)
Running the Cox PH model, using specified weights and step 2 data, between the treatments of interest. An HR was obtained for each IPD trial versus APD trial.
-
(4)
Performing a meta-analysis to obtain a final pooled HR of ozanimod versus DMF for the endpoint of interest (CDP at 3 months) using HRs from step 3.
It was not possible to obtain HRs for CDP at 6 months, since the CONFIRM and DEFINE studies did not report Kaplan–Meier curves at 6 months. Therefore, RRs using person-years as the denominator were used to assess CDP at 6 months. CDP RRs for APD imputed person-years by multiplying the number of people achieving CDP by total study duration, whereas time on study was used for IPD to calculate person-years. Imputing study duration for APD trials using total study duration is more conservative than other methods, such as using half of time on study.
3 Results
3.1 Baseline Characteristics Before and After Matching
Baseline patient characteristics from pooled DMF trials used to weight ozanimod trials, as well as unweighted and weighted IPD, are presented in Table 1, and the estimated sample size (ESS) for each outcome of interest is shown in Table 2. Region was not included as a prognostic factor for all outcomes because of nonconvergence (where the specified weight adjustments to the IPD did not result in a difference of 0 between weighted IPD and APD). Additionally, weight was excluded as a potential prognostic factor for the SUNBEAM trial in AE outcomes because of nonconvergence. Although the associated ESS for each outcome of interest was reduced after weighting, the relatively large sample indicated that the distribution of weights was not highly variable and the populations were sufficiently matched.
3.2 Adjusted Analyses
After adjustment for baseline patient characteristics, ozanimod 1.0 mg was associated with a decreased risk of relapse compared with DMF 240 mg, as evidenced by both a lower ARR (RR 0.80 [95% CI 0.67–0.97]) and lower proportion relapsed (OR 0.66 [95% CI 0.52–0.83]; Table 3). Compared with DMF 240 mg, ozanimod 1.0 mg was also associated with a significantly decreased risk of CDP at 3 months, accounting for underlying risk of CDP using a Cox PH model (HR 0.67 [95% CI 0.53–0.86]), but not at 6 months (RR 0.79 [95% CI 0.58–1.07]), regardless of time on study. Compared with patients receiving DMF 240 mg, patients receiving ozanimod 1.0 mg had a lower risk of experiencing an SAE (OR 0.27 [95% CI 0.19–0.39]), any AE (OR 0.11 [95% CI 0.08–0.16]), or discontinuation due to an AE (OR 0.11 [95% CI 0.07–0.17]).
4 Discussion
To date, no head-to-head clinical studies have compared ozanimod and DMF in patients with RMS to assess the relative clinical efficacy and safety of these two therapies. The current analysis provides an indirect comparison of ozanimod and DMF for treatment of RMS while adjusting for cross-trial differences to assess the efficacy and safety of these two treatments. MAIC methodology has been increasingly used to provide comparisons of RRMS therapies. Swallow et al. [50] conducted a MAIC of ozanimod and fingolimod and found that ozanimod was associated with a significantly lower incidence of AE outcomes at first-dose monitoring and over 1–2 years of follow-up, whereas the two agents had similar outcomes for the key endpoints of CDP and ARR. Fox et al. [48] conducted a MAIC of fingolimod and DMF to adjust for differences in baseline characteristics across the fingolimod (FREEDOMS/FREEDOMS II) and DMF (DEFINE/CONFIRM) trials to examine clinical measures of both relapse and disability. This MAIC indicated that fingolimod had efficacy similar to that of delayed-release DMF with respect to ARRs, 12-week CDP, evidence of disease activity, and the Multiple Sclerosis Functional Composite score [48]. Overall, these studies demonstrated that ozanimod was associated with an improved safety and similar efficacy profile compared with fingolimod, whereas DMF was not statistically differentiated from fingolimod with respect to relapse and CDP outcomes [48, 50].
The results of the current ozanimod–DMF MAIC adds to the evidence from previous studies, demonstrating the utility of MAICs of DMTs and may play an important role in assessing treatment choices for patients with RMS. To make reliable indirect comparisons, patient populations need to be as similar as possible in the absence of a common comparator arm and have cross-trial heterogeneity. When there is access to IPD for one treatment arm and APD for the other treatment arm, leveraging all available information allows an assessment of relative treatment outcomes for safety and efficacy endpoints, and this comparison is essential for filling in evidence gaps. This is especially important in a disease area such as RMS in which unmet treatment needs remain despite an evolving treatment landscape [5].
In the current analysis, ozanimod 1.0 mg was associated with a reduced risk of relapse and CDP at 3 months compared with DMF 240 mg, whereas the two treatments were comparable with respect to CDP at 6 months. However, CDP results are subject to uncertainty given the low absolute event rates observed for ozanimod in the SUNBEAM and RADIANCE-B trials, especially the 6-month CDP rate of only 5.8% at 2 years [57]. Moreover, patient characteristics in RRMS trials have changed, leading to inclusion of patients with milder disease in more recent trials [58]. Similar to other recent trials in RRMS, patients had baseline characteristics (mean age 36 years; mean time since symptom onset 6.7 years; mean EDSS 2.6) that reflected a population with mild to moderate disease. In addition, approximately 70% of patients in the ozanimod clinical trials were treatment naïve. These characteristics are consistent with those of a population that has a lower likelihood of sustained disability progression [58]. Furthermore, CDP at 5 months yields lower event rates as it is a more rigorous measure than the more commonly used 3-month CDP. Some patients, for instance, who exhibit 3-month CDP could subsequently experience clinical improvement and thus not qualify for 6-month CDP. Some patients may have attained 3-month CDP, but the trial ended before 6-month CDP could be measured. These factors may contribute to the lower number of patients reaching CDP and therefore reduces the statistical power to make inferences in trials with durations of 1–2 years. This limitation increases uncertainty in the estimation of relative effectiveness on CDP, especially confirmed after 6 months.
The MAIC of ozanimod 1.0 mg versus DMF 240 mg also showed that ozanimod was associated with significantly lower risks of SAEs, AEs, and discontinuations due to AEs compared with DMF. These findings suggest that ozanimod has a favorable benefit–risk profile compared with DMF for efficacy and safety outcomes over a 1- to 2-year study duration. Using MAIC methods maximizes available data on the treatment comparisons of interest, and, in this study, we were able to provide further evidence that ozanimod has improved efficacy and safety over DMF, along with fingolimod, in patients with RMS [50].
The assessment of cross-trial differences was used to obtain matched patient populations with respect to known treatment-effect modifiers and prognostic factors. Studies have shown the use of prior DMTs and female sex were more likely to be positively associated with relapses throughout the course of the disease [59]. The ozanimod-treated population was matched to the DMF population by increasing the weights of patients receiving ozanimod who had received prior DMT, had a longer duration of disease, and were female, while reducing the weighting of patients from Eastern Europe. Despite matching patient characteristics that may contribute to worse patient outcomes, ozanimod treatment was associated with an improved safety and efficacy profile compared with DMF.
Since no common comparator was available between ozanimod and DMF, the current analysis used an unanchored MAIC. While this adjusts for all known prognostic factors and treatment-effect modifiers, it potentially results in an unknown magnitude of residual bias from unobserved prognostic variables and effect modifiers. In this study, several factors could not be adjusted because of lack of data availability from the DMF trials, which included ethnicity and disease duration from symptom onset. Additionally, the screening criteria between the studies differed with respect to relapse history and time of GdE+ lesions, which may have introduced an imbalance between the two therapy cohorts due to unobserved characteristics.
Differences in study design, which cannot be adjusted for by using baseline patient characteristics, may also have contributed to an unknown magnitude of residual bias. Despite these limitations, several measures were taken to limit bias, and the analysis used a robust methodology that followed technical guidance from the UK National Institute for Health and Care Excellence [47, 60]. All adjustments in the current analysis were tailored to include only studies reporting the outcome of interest for each comparison, thereby lowering the risk of biased relative treatment-effect estimates. To assess the degree to which bias was reduced, comparisons of relative treatment effects from the MAICs were compared with relative treatment effects derived from an unadjusted indirect treatment comparison (i.e., “naïve” comparison), the comparisons of estimates from adjusted IPD and unadjusted IPD, as well as assessment of sample size reduction and distributions of weights, all suggesting that model specification was sufficient and yielded robust estimates in this study. Furthermore, weight distributions were largely centered at 0, and the use of standard errors produced from the robust sandwich estimator suggest that the estimates generated using MAIC methodology were robust. Although the MAIC adjusts for key treatment-effect modifiers and prognostic factors, the possibility of confounding due to unobserved baseline differences remains.
5 Conclusion
In the absence of head-to-head clinical trials, indirect treatment comparison methods provide additional comparative evidence to inform decision making. The findings of this MAIC suggest that, compared with DMF, ozanimod is associated with significantly improved relapse and safety outcomes, lowered 3-month CDP, and better safety outcomes. Although a MAIC adjusts for key treatment-effect modifiers and prognostic factors, results may be confounded by unobserved differences between trials and the low rate of disability progression. Future comparative efficacy studies will be valuable to validate the findings of this study and provide further evidence for clinicians to use when making treatment decisions.
References
Lublin FD, Reingold SC, Cohen JA, Cutter GR, Sorensen PS, Thompson AJ, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014;83(3):278–86.
Finkelsztejn A. Multiple sclerosis: overview of disease-modifying agents. Perspect Medicin Chem. 2014;6:65–72.
European Medicines Agency. Guideline on clinical investigation of medicinal products for the treatment of multiple sclerosis. European Medicines Agency, London, UK. 2015. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-clinical-investigation-medicinal-products-treatment-multiple-sclerosis_en-0.pdf. Accessed 25 June 2020.
Olek M, Howard J. Clinical presentation, course, and prognosis of multiple sclerosis in adults. UpToDate. 2019. https://www.uptodate.com/contents/clinical-presentation-course-and-prognosis-of-multiple-sclerosis-in-adults. Accessed 25 June 2020.
Giovannoni G, Butzkueven H, Dhib-Jalbut S, Hobart J, Kobelt G, Pepper G, et al. Brain health: time matters in multiple sclerosis. Mult Scler Relat Disord. 2016;9(Suppl 1):S5–48.
Lublin FD, Baier M, Cutter G. Effect of relapses on development of residual deficit in multiple sclerosis. Neurology. 2003;61(11):1528–32.
Doshi A, Chataway J. Multiple sclerosis, a treatable disease. Clin Med (Lond). 2017;17(6):530–6.
Gehr S, Kaiser T, Kreutz R, Ludwig W-D, Paul F. Suggestions for improving the design of clinical trials in multiple sclerosis—results of a systematic analysis of completed phase III trials. EPMA J. 2019;10(4):425–36.
Straus Farber R, Harel A, Lublin F. Novel agents for relapsing forms of multiple sclerosis. Annu Rev Med. 2016;67:309–21.
Fox RJ, Miller DH, Phillips JT, Hutchinson M, Havrdova E, Kita M, et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med. 2012;367(12):1087–97.
Gold R, Kappos L, Arnold DL, Bar-Or A, Giovannoni G, Selmaj K, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med. 2012;367(12):1098–107.
Wright K, Winkler MD, Newton BD, Sormani MP, Okuda DT. Patient outcomes influenced by reduced lymphocyte counts after dimethyl fumarate initiation. Neurol Neuroimmunol Neuroinflamm. 2017;4(6):e397.
Longbrake EE, Ramsbottom MJ, Cantoni C, Ghezzi L, Cross AH, Piccio L. Dimethyl fumarate selectively reduces memory T cells in multiple sclerosis patients. Mult Scler. 2016;22(8):1061–70.
Longbrake EE, Cantoni C, Chahin S, Cignarella F, Cross AH, Piccio L. Dimethyl fumarate induces changes in B- and T-lymphocyte function independent of the effects on absolute lymphocyte count. Mult Scler. 2018;24(6):728–38.
Lundy SK, Wu Q, Wang Q, Dowling CA, Taitano SH, Mao G, et al. Dimethyl fumarate treatment of relapsing–remitting multiple sclerosis influences B-cell subsets. Neurol Neuroimmunol Neuroinflamm. 2016;3(2):e211.
Wu Q, Wang Q, Mao G, Dowling CA, Lundy SK, Mao-Draayer Y. Dimethyl fumarate selectively reduces memory T cells and shifts the balance between Th1/Th17 and Th2 in multiple sclerosis patients. J Immunol. 2017;198(8):3069–80.
Li R, Rezk A, Ghadiri M, Luessi F, Zipp F, Li H, et al. Dimethyl fumarate treatment mediates an anti-inflammatory shift in B cell subsets of patients with multiple sclerosis. J Immunol. 2017;198(2):691–8.
Longbrake EE, Mao-Draayer Y, Cascione M, Zielinski T, Bame E, Brassat D et al. Dimethyl fumarate treatment shifts the immune environment toward an anti-inflammatory cell profile while maintaining protective humoral immunity. Mult Scler. 2020:1352458520937282.
Piroli GG, Manuel AM, Patel T, Walla MD, Shi L, Lanci SA, et al. Identification of novel protein targets of dimethyl fumarate modification in neurons and astrocytes reveals actions independent of Nrf2 stabilization. Mol Cell Proteomics. 2019;18(3):504–19.
Linker RA, Lee DH, Ryan S, van Dam AM, Conrad R, Bista P, et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain. 2011;134(Pt 3):678–92.
Parodi B, Rossi S, Morando S, Cordano C, Bragoni A, Motta C, et al. Fumarates modulate microglia activation through a novel HCAR2 signaling pathway and rescue synaptic dysregulation in inflamed CNS. Acta Neuropathol. 2015;130(2):279–95.
Gopal S, Mikulskis A, Gold R, Fox RJ, Dawson KT, Amaravadi L. Evidence of activation of the Nrf2 pathway in multiple sclerosis patients treated with delayed-release dimethyl fumarate in the phase 3 DEFINE and CONFIRM studies. Mult Scler. 2017;23(14):1875–83.
Huang H, Taraboletti A, Shriver LP. Dimethyl fumarate modulates antioxidant and lipid metabolism in oligodendrocytes. Redox Biol. 2015;5:169–75.
Scannevin RH, Chollate S, Jung MY, Shackett M, Patel H, Bista P, et al. Fumarates promote cytoprotection of central nervous system cells against oxidative stress via the nuclear factor (erythroid-derived 2)-like 2 pathway. J Pharmacol Exp Ther. 2012;341(1):274–84.
Chen H, Assmann JC, Krenz A, Rahman M, Grimm M, Karsten CM, et al. Hydroxycarboxylic acid receptor 2 mediates dimethyl fumarate’s protective effect in EAE. J Clin Investig. 2014;124(5):2188–92.
Kastrati I, Siklos MI, Calderon-Gierszal EL, El-Shennawy L, Georgieva G, Thayer EN, et al. Dimethyl fumarate inhibits the nuclear factor κB pathway in breast cancer cells by covalent modification of p65 protein. J Biol Chem. 2016;291(7):3639–47.
Carlström KE, Ewing E, Granqvist M, Gyllenberg A, Aeinehband S, Enoksson SL, et al. Therapeutic efficacy of dimethyl fumarate in relapsing–remitting multiple sclerosis associates with ROS pathway in monocytes. Nat Commun. 2019;10(1):3081.
Lückel C, Picard F, Raifer H, Campos Carrascosa L, Guralnik A, Zhang Y, et al. IL-17+ CD8+ T cell suppression by dimethyl fumarate associates with clinical response in multiple sclerosis. Nat Commun. 2019;10(1):5722.
Saida T, Yamamura T, Kondo T, Yun J, Yang M, Li J, et al. A randomized placebo-controlled trial of delayed-release dimethyl fumarate in patients with relapsing-remitting multiple sclerosis from East Asia and other countries. BMC Neurol. 2019;19(1):5.
Tecfidera [package insert]. Cambridge. MA: Biogen Inc.; 2020.
European Medicines Agency. Tecfidera EPAR summary for the public. European Medicines Agency, London, UK. 2014. https://www.ema.europa.eu/en/documents/product-information/tecfidera-epar-product-information_en.pdf. Accessed 16 Sept 2020.
Scott FL, Clemons B, Brooks J, Brahmachary E, Powell R, Dedman H, et al. Ozanimod (RPC1063) is a potent sphingosine-1-phosphate receptor-1 (S1P1) and receptor-5 (S1P5) agonist with autoimmune disease-modifying activity. Br J Pharmacol. 2016;173(11):1778–92.
Brinkmann V, Cyster JG, Hla T. FTY720: sphingosine 1-phosphate receptor-1 in the control of lymphocyte egress and endothelial barrier function. Am J Transplant. 2004;4(7):1019–25.
Chiba K. FTY720, a new class of immunomodulator, inhibits lymphocyte egress from secondary lymphoid tissues and thymus by agonistic activity at sphingosine 1-phosphate receptors. Pharmacol Ther. 2005;108(3):308–19.
Tsai HC, Nguyen K, Hashemi E, Engleman E, Hla T, Han MH. Myeloid sphingosine-1-phosphate receptor 1 is important for CNS autoimmunity and neuroinflammation. J Autoimmun. 2019;105:102290.
Liu G, Yang K, Burns S, Shrestha S, Chi H. The S1P1-mTOR axis directs the reciprocal differentiation of TH1 and regulatory T cells. Nat Immunol. 2010;11(11):1047–56.
Liao JJ, Huang MC, Goetzl EJ. Cutting edge: alternative signaling of Th17 cell development by sphingosine 1-phosphate. J Immunol. 2007;178(9):5425–8.
Garris CS, Wu L, Acharya S, Arac A, Blaho VA, Huang Y, et al. Defective sphingosine 1-phosphate receptor 1 (S1P1) phosphorylation exacerbates TH17-mediated autoimmune neuroinflammation. Nat Immunol. 2013;14(11):1166–72.
Lee H, Deng J, Kujawski M, Yang C, Liu Y, Herrmann A, et al. STAT3-induced S1PR1 expression is crucial for persistent STAT3 activation in tumors. Nat Med. 2010;16(12):1421–8.
van Doorn R, Lopes Pinheiro MA, Kooij G, Lakeman K, van het Hof B, van der Pol SM, et al. Sphingosine 1-phosphate receptor 5 mediates the immune quiescence of the human brain endothelial barrier. J Neuroinflamm. 2012;9:133.
Nystad AE, Lereim RR, Wergeland S, Oveland E, Myhr KM, Bø L, et al. Fingolimod downregulates brain sphingosine-1-phosphate receptor 1 levels but does not promote remyelination or neuroprotection in the cuprizone model. J Neuroimmunol. 2020;339:577091.
Jaillard C, Harrison S, Stankoff B, Aigrot MS, Calver AR, Duddy G, et al. Edg8/S1P5: an oligodendroglial receptor with dual function on process retraction and cell survival. J Neurosci. 2005;25(6):1459–69.
Rasche L, Paul F. Ozanimod for the treatment of relapsing remitting multiple sclerosis. Expert Opin Pharmacother. 2018;19(18):2073–86.
Bristol Myers Squibb. Zeposia [package insert]. Princeton: Bristol Myers Squibb; 2020.
Cohen JA, Comi G, Selmaj KW, Bar-Or A, Arnold DL, Steinman L, et al. Safety and efficacy of ozanimod versus interferon beta-1a in relapsing multiple sclerosis (RADIANCE): a multicentre, randomised, 24-month, phase 3 trial. Lancet Neurol. 2019;18(11):1021–33.
Comi G, Kappos L, Selmaj KW, Bar-Or A, Arnold DL, Steinman L, et al. Safety and efficacy of ozanimod versus interferon beta-1a in relapsing multiple sclerosis (SUNBEAM): a multicentre, randomised, minimum 12-month, phase 3 trial. Lancet Neurol. 2019;18(11):1009–20.
Phillippo DM, Ades AE, Dias S, Palmer S, Abrams KR, Welton NJ. Methods for population-adjusted indirect comparisons in health technology appraisal. Med Decis Making. 2018;38(2):200–11.
Fox RJ, Chan A, Zhang A, Xiao J, Levison D, Lewin JB, et al. Comparative effectiveness using a matching-adjusted indirect comparison between delayed-release dimethyl fumarate and fingolimod for the treatment of multiple sclerosis. Curr Med Res Opin. 2017;33(2):175–83.
Berardi A, Siddiqui MK, Treharne C, Harty G, Wong SL. Estimating the comparative efficacy of cladribine tablets versus alternative disease modifying treatments in active relapsing–remitting multiple sclerosis: adjusting for patient characteristics using meta-regression and matching-adjusted indirect treatment comparison approaches. Curr Med Res Opin. 2019;35(8):1371–8.
Swallow E, Patterson-Lomba O, Yin L, Mehta R, Pelletier C, Kao D, et al. Comparative safety and efficacy of ozanimod versus fingolimod for relapsing multiple sclerosis. J Comp Eff Res. 2020;9(4):275–85.
Signorovitch JE, Sikirica V, Erder MH, Xie J, Lu M, Hodgkins PS, et al. Matching-adjusted indirect comparisons: a new tool for timely comparative effectiveness research. Value Health. 2012;15(6):940–7.
Signorovitch JE, Wu EQ, Yu AP, Gerrits CM, Kantor E, Bao Y, et al. Comparative effectiveness without head-to-head trials: a method for matching-adjusted indirect comparisons applied to psoriasis treatment with adalimumab or etanercept. Pharmacoeconomics. 2010;28(10):935–45.
Prosperini L, Lucchini M, Haggiag S, Bellantonio P, Bianco A, Buscarinu MC, et al. Fingolimod vs. dimethyl fumarate in multiple sclerosis: a real-world propensity score-matched study. Neurology. 2018;91(2):e153–61.
Ishak KJ, Rael M, Hicks M, Mittal S, Eatock M, Valle JW. Relative effectiveness of sunitinib versus everolimus in advanced pancreatic neuroendocrine tumors: an updated matching-adjusted indirect comparison. J Comp Eff Res. 2018;7(10):947–58.
Guyot P, Ades AE, Ouwens MJ, Welton NJ. Enhanced secondary analysis of survival data: reconstructing the data from published Kaplan–Meier survival curves. BMC Med Res Methodol. 2012;12:9.
DigitizeIt. Digitizer software—digitize a scanned graph or chart into (x,y)-data. DigitizeIt. 2020. https://www.digitizeit.de/. Accessed 02 July 2020.
Zeposia [summary of product characteristics]. Utrecht, Netherlands: Celgene Distribution B.V.; 2020.
Zhang Y, Salter A, Wallstrom E, Cutter G, Stuve O. Evolution of clinical trials in multiple sclerosis. Ther Adv Neurol Disord. 2019;12:1756286419826547.
Kalincik T. Multiple sclerosis relapses: epidemiology, outcomes and management: a systematic review. Neuroepidemiology. 2015;44(4):199–214.
Phillippo DM, Ades AE, Dias S, Palmer S, Abrams KR, Welton NJ. NICE DSU Technical Support Document 18: Methods for Population-Adjusted Indirect Comparisons in Submissions to NICE. National Institute for Health and Care Excellence Decision Support Unit, Sheffield, UK. 2016. http://nicedsu.org.uk/wp-content/uploads/2018/08/Population-adjustment-TSD-FINAL-ref-rerun.pdf. Accessed 25 June 2020.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was supported by Bristol Myers Squibb, Princeton, NJ. Bristol Myers Squibb was involved in the design and conduct of the study; collection, management, analysis, and interpretation of the data; and preparation, review, and approval of the manuscript. Publication fees were paid by Bristol Myers Squibb. The authors directed and are fully responsible for all content and editorial decisions for this manuscript.
Conflict of interest
SC: Advisory board – AbbVie, Biogen, Novartis, Pear Therapeutics, Roche Genentech, and Sanofi Genzyme; speaker – Biogen, Novartis, Roche Genentech, and Sanofi Genzyme; grant/research support to SC employer Providence Brain and Spine Institute – AbbVie, Adamas, Altios, Biogen, EMD Serono, MedDay, NARCRMS, Novartis, Providence St. Vincent Foundation, Roche Genentech, and Sanofi Genzyme; consultant – AbbVie, Bristol Myers Squibb. SA, XZ & MZ: Employment – Genesis Research (Genesis Research provided consulting services to Celgene, a Bristol Myers Squibb). TT & JK: Employment –a Bristol Myers Squibb company.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Data requests may be submitted to Celgene, a Bristol Myers Squibb company, at https://vivli.org/ourmembers/celgene.
Code availability
Not applicable.
Author contributions
MZ, SA, and XZ were responsible for the study design and data analysis. All authors were the study investigators, interpreted the data, and prepared, reviewed, revised, and gave approval for publication of the manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Cohan, S., Kumar, J., Arndorfer, S. et al. Comparative Efficacy and Safety of Ozanimod and Dimethyl Fumarate for Relapsing-Remitting Multiple Sclerosis Using Matching-Adjusted Indirect Comparison. CNS Drugs 35, 795–804 (2021). https://doi.org/10.1007/s40263-021-00805-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-021-00805-0