
Abstract
Objectives
The aim of this study was to compare the efficacy and safety of the prodrug psychostimulant lisdexamfetamine dimesylate (LDX) and the non-stimulant noradrenergic compound atomoxetine (ATX) in children and adolescents with attention-deficit/hyperactivity disorder (ADHD) who had previously responded inadequately to methylphenidate (MPH).
Methods
This 9-week, head-to-head, randomized, double-blind, active-controlled study (SPD489-317; ClinicalTrials.gov NCT01106430) enrolled patients (aged 6–17 years) with at least moderately symptomatic ADHD and an inadequate response to previous MPH therapy. Patients were randomized (1:1) to an optimized daily dose of LDX (30, 50 or 70 mg) or ATX (patients <70 kg, 0.5–1.2 mg/kg with total daily dose not to exceed 1.4 mg/kg; patients ≥70 kg, 40, 80 or 100 mg). The primary efficacy outcome was time (days) to first clinical response. Clinical response was defined as a Clinical Global Impressions-Improvement (CGI-I) score of 1 (very much improved) or 2 (much improved). Secondary efficacy outcomes included the proportion of responders at each study visit and the change from baseline in ADHD Rating Scale (ADHD-RS-IV) and CGI-Severity scores. Tolerability and safety were assessed by monitoring treatment-emergent adverse events (TEAEs), height and weight, vital signs and electrocardiogram parameters. Endpoint was defined as the last post-baseline, on-treatment visit with a valid assessment.
Results
Of 267 patients randomized (LDX, n = 133; ATX, n = 134), 200 (74.9 %) completed the study. The median time to first clinical response [95 % confidence interval (CI)] was significantly shorter for patients receiving LDX [12.0 days (8.0–16.0)] than for those receiving ATX [21.0 days (15.0–23.0)] (p = 0.001). By week 9, 81.7 % (95 % CI 75.0–88.5) of patients receiving LDX had responded to treatment compared with 63.6 % (95 % CI 55.4–71.8) of those receiving ATX (p = 0.001). Also by week 9, the difference between LDX and ATX in least-squares mean change from baseline (95 % CI) was significant in favour of LDX for the ADHD-RS-IV total score [−6.5 (−9.3 to −3.6); p < 0.001; effect size 0.56], inattentiveness subscale score [−3.4 (−4.9 to −1.8); p < 0.001; effect size 0.53] and the hyperactivity/impulsivity subscale score [−3.2 (−4.6 to −1.7); p < 0.001; effect size 0.53]. TEAEs were reported by 71.9 and 70.9 % of patients receiving LDX and ATX, respectively. At endpoint, both treatments were associated with mean (standard deviation) increases in systolic blood pressure [LDX, +0.7 mmHg (9.08); ATX, +0.6 mmHg (7.96)], diastolic blood pressure [LDX, +0.1 mmHg (8.33); ATX, +1.3 mmHg (8.24)] and pulse rate [LDX, +3.6 bpm (10.49); ATX, +3.7 bpm (10.75)], and decreases in weight [LDX, −1.30 kg (1.806); ATX, −0.15 kg (1.434)].
Conclusions
LDX was associated with a faster and more robust treatment response than ATX in children and adolescents with at least moderately symptomatic ADHD who had previously responded inadequately to MPH. Both treatments displayed safety profiles consistent with findings from previous clinical trials.
Similar content being viewed by others

1 Introduction
Attention-deficit/hyperactivity disorder (ADHD) is estimated to affect 5 % of children worldwide, making it one of the most common neurodevelopmental disorders in children and adolescents [1]. There are considerable differences in the worldwide availability of medications for ADHD. In North America, methylphenidate (MPH)- and amfetamine-based psychostimulants are considered first-line ADHD therapies, and a variety of short- and long-acting formulations are available [2, 3]. In Europe, short- and long-acting MPH formulations are widely available, and MPH is generally considered to be the first-line medication for ADHD [4]. Until recently, only short-acting formulations of amfetamine have been approved in Europe, and only in some countries [4]. Of non-stimulant ADHD medications, the noradrenaline reuptake inhibitor atomoxetine (ATX) is approved in North America and several European countries [5], and the α2-adrenergic agonists clonidine and guanfacine are licensed in the USA but not Europe [6, 7].
Lisdexamfetamine dimesylate (LDX) is a long-acting prodrug treatment for patients with ADHD [8]. Multiple randomized, double-blind, placebo-controlled trials have shown LDX to be an effective treatment for children, adolescents and adults with ADHD, with a tolerability and safety profile consistent with the known effects of psychostimulant therapy [9–15]. LDX is approved as a first-line treatment for ADHD in the USA, Canada and Brazil. In early 2013, LDX became the first long-acting, amfetamine-based medication to be approved in Europe, where it is licensed in several countries for the treatment of children and adolescents with ADHD who have had a clinically inadequate response to MPH. Here, we present results from a head-to-head, 9-week, double-blind, randomized, active-controlled, phase IIIb study (SPD489-317; ClinicalTrials.gov NCT01106430). This study was designed to provide a direct comparison of the efficacy and safety of LDX and ATX in children and adolescents with ADHD who had experienced an inadequate response to previous MPH therapy. These results will aid clinicians when developing individualized treatment plans for the management of patients with ADHD.
2 Methods
This double-blind, randomized, active-controlled, parallel-group study was approved by an independent ethics committee/institutional review board and regulatory agency in each centre (as appropriate). The study was conducted in accordance with current applicable regulations, the International Conference on Harmonization of Good Clinical Practice [16] and local ethical and legal requirements. Before enrolment in the study, written, informed consent was obtained from the necessary parent(s) or legal guardian(s) for each patient, in accordance with local requirements, and assent was also obtained from each patient, when applicable.
2.1 Study Population
This study enrolled male and female patients (aged 6–17 years) who satisfied Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DSM-IV-TR) [17] criteria for a primary diagnosis of ADHD of at least moderate severity as shown by a baseline ADHD Rating Scale IV (ADHD-RS-IV) total score of 28 or higher. Inclusion and exclusion criteria related to a patient’s previous exposure and/or response to ADHD medications are outlined in Table 1. Other inclusion criteria included age-appropriate intellectual functioning; ability to swallow a capsule; and blood pressure measurements within the 95th percentile for age, sex and height. Female patients of childbearing potential were required to have a negative urine pregnancy test at baseline and to comply with the contraceptive requirements of the protocol. Other key exclusion criteria included comorbid psychiatric diagnosis with significant symptoms (based on Kiddie-Schedule for Affective Disorders and Schizophrenia for School Age Children—Present and Lifetime diagnostic interview); conduct disorder (excluding oppositional defiant disorder); suicide risk, with a previous suicide attempt or active suicidal ideation; pregnancy or lactation; weight below 22.7 kg; body mass index (BMI, kg/m2) greater than the 97th percentile for age and sex; positive urine drug test (with the exception of a patient’s current ADHD medication); clinically significant electrocardiogram (ECG) results or laboratory abnormalities; known CYP2D6 poor-metabolizer genotype; suspected substance abuse or dependence disorder (excluding nicotine) within the previous 6 months; history of seizures; tics or Tourette’s disorder; pre-existing liver disease or laboratory evidence of liver disease; known cardiac structural abnormality; or any other condition that might increase vulnerability to the sympathomimetic effects of a psychostimulant drug.
2.2 Study Design
Patients were required to discontinue any psychoactive medication for a 7-day washout period prior to baseline (visit 0). At baseline, patients were randomized (1:1) to receive a once-daily, morning dose (at 07:00 ± 2 h) of LDX or ATX for a 9-week, double-blind evaluation period (Fig. 1) with weekly, on-site efficacy, tolerability and safety assessments. Dosing began on the morning after the baseline visit and continued for 9 weeks, starting with a 4-week, stepwise, dose-optimization stage. Randomization of patients was stratified by country, and an automated interactive response system was used to generate the random (concealed) allocation sequence and assign participants to study treatments; patients, caregivers and investigators were blinded to the treatment allocation. All study drugs were over-encapsulated so they appeared identical.

Study design. Visit window ±2 days throughout the evaluation period. Visit window +2 days for safety follow-up visit. ATX atomoxetine, ET early termination, LDX lisdexamfetamine dimesylate, V visit
The dose-optimization phase involved adjustment of the dose until an ‘acceptable’ response was achieved [defined as a reduction of at least 30 % from baseline in the ADHD-RS-IV total score and a Clinical Global Impressions-Improvement (CGI-I) score of 1 or 2 with tolerable side effects]. Only one dose reduction was permitted during the optimization phase and, following dose reduction, further increases were not allowed. Dose adjustments were not permitted beyond visit 3, and patients who were unable to tolerate the study drug were withdrawn from the study.
LDX was provided in a single capsule of 30, 50 or 70 mg, with patients initially receiving a 30-mg dose. ATX was available in 10-, 18-, 25-, 40- and 60-mg capsules. All patients in the ATX group who weighed less than 70 kg were started on a daily dose of approximately 0.5 mg/kg body weight, the final target daily dose being 1.2 mg/kg, with a maximum permitted daily dose of 1.4 mg/kg. Patients who weighed 70 kg or more initially received 40 mg and, if required, were titrated to 80 mg and then to 100 mg daily. Some patients treated with ATX would need two capsules to achieve the required dose (e.g. 80 and 100 mg were achieved using two capsules). Therefore, all patients weighing more than 64.5 kg who were titrated to a higher dose were instructed to take two capsules (the second capsule could be either active drug or placebo, as appropriate) to maintain the double-blind study design.
2.3 Efficacy
The primary efficacy measure was the time to first clinical response (days) after the initiation of treatment, as assessed by CGI-I scores [18]. The CGI scale provides a global assessment of a patient’s severity of illness; CGI-I scores, which were reported at each post-baseline visit, rate the change in a patient’s condition from baseline on a scale of 1 (very much improved) to 7 (very much worse) [18]. Clinical ‘response’ was defined as a CGI-I score of 1 or 2 (very much improved or much improved). The number of days to first clinical response was calculated as the date of response minus the date of first dose, plus 1 day.
Secondary efficacy outcomes included the proportion of CGI-I responders at each study visit, the proportion of patients who had a decrease of at least one CGI-Severity (CGI-S) category from baseline (assessed at visit 4 and at visit 9), and the change from baseline in ADHD-RS-IV total and subscale scores at each study visit. CGI-S scores, which rate the severity of a patient’s condition on a scale of 1 (normal, not at all ill) to 7 (among the most extremely ill), were assessed at baseline, visit 4 and visit 9 [18]. The ADHD-RS-IV scale [19], designed to reflect current ADHD symptoms, assesses 18 items on a scale of 0 (no symptoms) to 3 (severe symptoms), with a total score ranging from 0 to 54. CGI and ADHD-RS-IV assessments were performed by a medical practitioner or psychologist experienced in the evaluation of children and adolescents with ADHD.
2.4 Tolerability and Safety
Tolerability and safety assessments included the monitoring of treatment-emergent adverse events (TEAEs), laboratory evaluations, physical examination (including weight), and monitoring of vital signs and ECG parameters. TEAEs were defined as adverse events that first occurred or worsened during the time between the first dose of study drug and the third day following cessation of treatment (inclusive). All TEAEs were coded using Medical Dictionary for Regulatory Activities (MedDRA) (version 14.1) [20]. A serious TEAE was any untoward medical occurrence that resulted in death; was life-threatening; required inpatient hospitalization or prolonged existing hospitalization; resulted in persistent or significant disability/incapacity; was a congenital abnormality/birth defect; or was an Important Medical Event. Important Medical Events may have been considered as serious TEAEs when, based upon medical judgement, they may have jeopardized the patient and may have required medical or surgical intervention to prevent one of the outcomes listed above. The sponsor required any new onset of seizures, syncope or loss of consciousness to be reported as a serious TEAE. Using Centers for Disease Control and Prevention percentile growth charts [21], BMI was categorized into five groups based on BMI percentiles: underweight (<5 %), healthy weight (low; ≥5 to <25 %), healthy weight (high; ≥25 to <85 %), at risk of being overweight (≥85 to <95 %) and overweight (≥95 %). ECG parameters were assessed at screening and visit 4. However, the visit 4 ECG was added as a result of a protocol amendment requested by the French Central Ethics Committee and therefore was not obtained for all patients. The Brief Psychiatric Rating Scale for Children (BPRS-C), the Columbia-Suicide Severity Rating Scale (C-SSRS) and the Udvalg for Kliniske Undersøgelser Side Effect Rating Scale-Clinician (UKU-SERS-Clin) were also used to monitor patient tolerability and safety plus the suitability of individuals to remain in the study [22–24].
2.5 Statistical Analyses
Safety/tolerability assessments were performed using the safety population, defined as all patients who were randomized and received at least one dose of study drug. Efficacy data were analysed using the full analysis set (FAS), also defined as all patients who were randomized and received at least one dose of study drug. One patient was randomized to ATX but received LDX owing to a drug dispensing error. Based on the intent-to-treat principle, this patient was included in the ATX treatment group in the FAS, but was counted in the LDX treatment group in the safety population. Endpoint was defined as the last on-treatment, post-baseline visit with a valid assessment.
Time to first clinical response (days) was calculated using Kaplan–Meier estimates and analysed using a Peto–Peto–Prentice–Wilcoxon (PPPW) test [25, 26], stratified by country and evaluated at a significance level of 0.05 (two-sided). The null hypothesis stated that there was no difference in the time to first clinical response between patients receiving LDX and those taking ATX, with the two-sided alternative of a non-zero difference between the groups. Allowing for a 20 % discontinuation rate, approximately 262 patients (131 in each treatment group) were required to detect a difference in time to first clinical response between the treatment groups with a power of 85 %. Patients who prematurely discontinued from the study without responding, and patients who completed the study up to visit 9 without meeting response criteria, were censored at visit 9 in the primary analysis of time to response, and classified as non-responders in the analysis of responders.
The proportion of responders (CGI-I score of 1 or 2) at each study visit and the proportion of patients who had a decrease of at least one CGI-S category by visit 9 were assessed using the last-observation-carried-forward (LOCF) approach and analysed using a Cochran–Mantel–Haenszel (CMH) test stratified by country. At each study visit, the change from baseline in ADHD-RS-IV scores, using LOCF, was analysed using an analysis of covariance (ANCOVA) model including treatment group (effect of interest), country (blocking factor) and the corresponding baseline score (covariate). Effect sizes were calculated as the difference in least-squares (LS) mean score between the two treatments, divided by the root mean square error obtained from the ANCOVA model. Effect sizes of 0.2, 0.5 and 0.8 correspond to small, medium and large magnitudes of effect, respectively [27].
3 Results
3.1 Patient Disposition and Baseline Characteristics
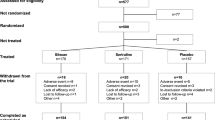
This study, conducted between 28 June 2010 and 19 July 2012, enrolled 267 patients from 51 sites in Canada (n = 35 patients), the USA (n = 138) and seven European countries (Belgium, n = 2; Germany, n = 42; Hungary, n = 20; Italy, n = 1; Poland, n = 1; Spain, n = 22; and Sweden, n = 6). Of 267 patients randomized (LDX, n = 133; ATX, n = 134), 200 (74.9 %) completed the study (LDX, n = 99; ATX, n = 101) (Fig. 2). The safety population comprised all patients who were randomized and received at least one dose of study drug (LDX, n = 128; ATX, n = 134). Based on the intent-to-treat principle, the FAS comprised 262 patients (LDX, n = 127; ATX, n = 135). The two most commonly reported reasons for study discontinuation for patients receiving LDX were adverse events (8/133 patients; 6.0 %) and withdrawal by patient (8/133; 6.0 %), and for patients receiving ATX they were lack of efficacy (13/134 patients; 9.7 %) and adverse events (10/134; 7.5 %). Baseline characteristics were similar for both treatment groups (Table 2).

Patient disposition. aThe safety population included all patients who were randomized and received at least one dose of study drug. bThe FAS included all patients who were randomized and received at least one dose of study drug. One patient was randomized to ATX but, owing to a drug dispensing error, received LDX. Based on the intent-to-treat principle, this patient was included in the ATX treatment group in the FAS. cStudy completers were patients who completed visits 0–10 (visit 10 being a clinic visit or telephone call). dOther reasons for discontinuation among patients administered LDX were difficulty swallowing capsule (n = 1); early termination requested by the sponsor because of previous marijuana use (n = 1); and early termination requested by the sponsor because patient was unable to meet the study visit schedule (n = 1). eOther reasons for discontinuation among patients administered ATX were refusal to take medication (n = 1); patient relocation due to a family emergency (n = 1); and non-compliance (n = 1). ATX atomoxetine, FAS full analysis set, LDX lisdexamfetamine dimesylate
3.2 Dose Optimization
The mean optimal dose (which was the dose that was dispensed at visit 4) for patients who received LDX during the dose-maintenance phase was 52.5 mg/day [standard deviation (SD): 16.10]; 28/128 (21.9 %) received 30 mg/day, 36/128 (28.1 %) received 50 mg/day and 41/128 (32.0 %) received 70 mg/day. The mean optimal dose for patients who received ATX was 40.2 mg/day (SD: 20.05); 15/134 (11.2 %) and 95/134 (70.9 %) received a daily dose of 0.5 mg/kg and 1.2 mg/kg, respectively (patients weighing <70 kg; n = 127), and 2/134 (1.5 %), 1/134 (0.7 %) and 4/134 (3.0 %) received 40, 80 and 100 mg/day, respectively (patients weighing ≥70 kg; n = 7). An optimal dose was not available for patients who discontinued the study before reaching visit 4 (LDX, n = 23; ATX, n = 17).
3.3 Efficacy
The mean CGI-S score at baseline was 5.0 in both treatment groups (Table 2), with most patients categorized as moderately ill or markedly ill (LDX, 97/127; ATX, 104/135). The median time to first clinical response (CGI-I score of 1 or 2) was significantly shorter for patients receiving LDX [12.0 days (95 % confidence interval [CI] 8.0–16.0)] than those receiving ATX [21.0 days (15.0–23.0); p = 0.001]. Significantly greater proportions of patients receiving LDX than of those receiving ATX responded to treatment at each study visit (all p < 0.01) (Fig. 3). By visit 9, 81.7 % (95 % CI 75.0–88.5) of patients receiving LDX had responded compared with 63.6 % (55.4–71.8) of those receiving ATX (p = 0.001).

Proportion of patients with a clinical response to LDX or ATX treatment (defined as a CGI-I score of 1 or 2) at each weekly study visit using LOCF. Values are shown as the proportion of responders ±95 % confidence intervals based on LOCF. ***p < 0.001, **p < 0.01 based on a Cochran–Mantel–Haenszel test stratified by country comparing LDX treatment with ATX. ATX atomoxetine, CGI-I Clinical Global Impressions-Improvement, LDX lisdexamfetamine dimesylate, LOCF last-observation-carried-forward
The proportion of patients with a decrease of at least one category from baseline in CGI-S score was significantly greater in the LDX treatment group than in the ATX treatment group by visit 4 [LDX, 92.3 % (95 % CI 87.5–97.1); ATX, 81.3 % (74.4–88.2); p < 0.05] and by visit 9 [LDX, 92.3 % (87.5–97.1); ATX, 79.7 % (72.6–86.8); p < 0.01].
There was no difference in mean (SD) ADHD-RS-IV total score at baseline between treatment groups (Table 2). Reductions from baseline in mean ADHD-RS-IV total scores were observed in both treatment groups; by visit 9, the mean (SD) ADHD-RS-IV total score was 16.3 (11.16) in the LDX group and 22.5 (13.21) in the ATX group. The mean (SD) change from baseline in ADHD-RS-IV total score by visit 9 was −26.3 (11.94) in the LDX group and −19.4 (12.82) in the ATX group. However, LDX treatment was associated with significantly greater reductions from baseline than ATX treatment (p < 0.001 for each study visit) (Fig. 4). By visit 9, the difference between LDX and ATX in LS mean change (95 % CI) from baseline was −6.5 (−9.3 to −3.6), with an effect size of 0.56. In addition, the differences (LDX minus ATX) in LS mean change from baseline (95 % CI) by visit 9 were statistically significant in favour of LDX (p < 0.001) for both the inattentiveness subscale [−3.4 (−4.9 to −1.8); effect size 0.53] and the hyperactivity/impulsivity subscale [−3.2 (−4.6 to −1.7); effect size 0.53].

Mean ADHD-RS-IV total scores (using LOCF) in patients treated with LDX or ATX. Mean ADHD-RS-IV total scores are shown ±95 % confidence intervals based on LOCF. ***p < 0.001 for LDX treatment compared with ATX based on an ANCOVA model of the least-squares mean change from baseline including treatment group (effect of interest), country (blocking factor) and the corresponding baseline total score (covariate). ADHD attention-deficit/hyperactivity disorder, ADHD-RS-IV ADHD Rating Scale IV, ANCOVA analysis of covariance, ATX atomoxetine, LDX lisdexamfetamine dimesylate, LOCF last-observation-carried-forward
3.4 Tolerability and Safety
TEAEs were reported by 92/128 patients (71.9 %) receiving LDX and 95/134 patients (70.9 %) receiving ATX (Table 3). Most TEAEs were mild to moderate in severity and no deaths or serious TEAEs were reported. The TEAEs that led to discontinuation of LDX were agitation, decreased weight, excoriation, indifference, irritability, nausea, somnolence and tic. The TEAEs leading to discontinuation of ATX were headache, irritability, epigastric discomfort, fatigue, influenza, malaise, nausea, sedation, somnolence and upper abdominal pain.
At endpoint, both LDX and ATX were associated with modest increases in mean systolic blood pressure, diastolic blood pressure and pulse rate (Table 4). At any point while on treatment, 4/127 patients (3.1 %) receiving LDX, and no patients receiving ATX, were classified as having a low pulse rate (defined as ≤50 bpm), whereas 19/127 (15.0 %) receiving LDX and 32/132 (24.2 %) receiving ATX met the outlier criterion for high pulse rate (defined as ≥100 bpm). Similar proportions of children (aged 6–12 years) experienced high systolic blood pressure [defined as >120 mmHg; LDX, 12/94 (12.8 %); ATX, 11/98 (11.2 %)], or diastolic blood pressure [defined as >80 mmHg; LDX, 11/94 (11.7 %); ATX, 13/98 (13.3 %)]. While no adolescent patients (aged 13–17 years) met the predefined outlier criterion for high systolic (>140 mmHg) or diastolic (>90 mmHg) blood pressure, supportive analyses indicated that the proportions of adolescents with systolic blood pressure >130 mmHg at any point during treatment were 2/33 (6.1 %) for LDX and 3/34 (8.8 %) for ATX; or for >120 mmHg were 20/33 (60.6 %) for LDX and 16/34 (47.1 %) for ATX; and for diastolic blood pressure >80 mmHg were 7/33 (21.2 %) for LDX and 6/34 (17.6 %) for ATX. No patients withdrew from the study as a result of a clinically significant blood pressure or pulse rate measurement.
The mean (SD) change in weight from baseline to endpoint was greater for patients receiving LDX [−1.30 kg (1.806)] than ATX [−0.15 kg (1.434)] (Table 4), and the outlier criterion for weight reduction (defined as ≥7 % reduction from baseline) was met by more patients receiving LDX [34/127 (26.8 %)] than ATX [6/132 (4.5 %)]. At endpoint, shifts to lower BMI categories were reported for 19/127 patients receiving LDX and 15/132 on ATX, whereas 4/127 patients on LDX and 7/132 on ATX had shifted to a higher BMI category. Five patients moved into the underweight category (LDX, n = 4; ATX, n = 1); all five were in the healthy weight (low) category at baseline and all completed the study.
ECG parameters, assessed at screening and visit 4, are shown in Table 4. In both treatment groups, some patients experienced potentially clinically important (PCI) readings for heart rate [defined as ≥100 bpm; LDX, 8/83 (9.6 %); ATX, 8/91 (8.8 %)], PR interval [defined as ≥200 ms; LDX, 0; ATX, 1/91 (1.1 %)] and QTcF (QT interval corrected using Fridericia’s formula) change from screening [defined as ≥30 or <60 ms; LDX, 2/83 (2.4 %); ATX, 1/90 (1.1 %)]. No patients experienced a PCI QTcF absolute reading (defined as ≥450 ms), and no patients withdrew from the study as a result of a clinically significant ECG measurement.
4 Discussion
In this direct comparison between LDX and ATX, both treatments led to improvements in symptoms and behaviours associated with ADHD in children and adolescents who had previously experienced an inadequate response to MPH therapy. However, the efficacy of LDX was significantly faster to onset and greater than that of ATX. Both therapies displayed safety profiles consistent with findings from previous studies [9, 11–13, 28–30]. These results support previous placebo-controlled trials of LDX, which have demonstrated a robust treatment response in a range of patient groups [9, 11–13].
In the current study, patients were required to have experienced an inadequate response to previous MPH therapy. In most cases, this was due to a lack of efficacy of MPH (LDX, 75.6 %; ATX, 78.5 %). Despite this, the majority of patients receiving LDX (81.7 %) and ATX (63.6 %) were classified as treatment responders by visit 9, according to CGI-I criteria. These results are of particular relevance to the recent approval of LDX in Europe for the treatment of children and adolescents whose response to previous MPH treatment is considered clinically inadequate. The observation of a robust LDX treatment response in patients previously treated with MPH is supported by a post hoc analysis of a randomized, double-blind, US-based study in 290 children with ADHD [31]. That study reported no difference in response to LDX between the overall study population and the subgroup of patients who were receiving MPH at study entry but were not considered well controlled. In addition, several studies have demonstrated that, although the overall response to MPH and amfetamine is similar, the response to each varies among individual patients, and non-response to one class of psychostimulant does not predict non-response to a second [32].
A subgroup analysis of a double-blind, placebo-controlled trial in 125 children with ADHD concluded that the effects of ATX treatment were largely independent of previous exposure to psychostimulants [33]. Based on ADHD-RS-IV total scores, effect sizes relative to placebo were 0.75 in pre-treated patients and 0.97 in patients who had not received previous psychostimulant treatment (interaction with treatment, p = 0.607) [33]. In support of this conclusion, a meta-analysis including six randomized controlled trials found that previous treatment with psychostimulants did not influence clinical response to ATX [34]. In contrast, one large (n = 516), double-blind, placebo-controlled, crossover study found that the proportions of stimulant-naïve children with ADHD who responded to osmotic-release oral system MPH (OROS-MPH) and ATX (64 and 57 %, respectively; effect sizes relative to placebo based on ADHD-RS-IV total score, 1.0 and 0.9) were higher than among those who had previously been treated with a psychostimulant (51 and 37 %, respectively; effect sizes 0.8 and 0.5) [35]. It is unclear why there are discrepancies between studies with respect to the impact of previous psychostimulant treatment on ATX response, but potential differences in the baseline severity of symptoms between previously treated and treatment-naïve patients may confound outcomes [33]. Overall, these data support the importance of alternative treatment options for patients who have not improved satisfactorily on their current ADHD medication.
The observed shorter time to first clinical response (CGI-I of 1 or 2) for LDX relative to ATX was not unexpected as psychostimulants are generally recognized to produce immediate treatment effects [3, 4]. In contrast, estimates of the time required for ATX to reach its maximum effect generally range from 4 to 6 weeks [3, 4], although, one study suggests that it may take as long as 12 weeks [36]. The mechanism that underlies this delay in onset of action of ATX is not known. The dose-optimized design of this study is likely to have had some influence on the time to first clinical response in both treatment arms. Importantly, however, as the dose-titration schedules for both treatments followed current clinical guidelines, the times to treatment response observed in this study are relevant to clinical practice.
In addition to a faster onset of efficacy, the proportions of patients who responded to treatment (CGI-I of 1 or 2) or improved by at least one CGI-S category, and the improvements in mean ADHD-RS-IV total and subscale scores, all indicated that there was a greater reduction of symptoms in patients receiving LDX than ATX by visit 9. A meta-analysis of 32 clinical trials and a total of 15 ADHD medications supports these observations, concluding that both short- and long-acting psychostimulants were significantly more effective than non-stimulants [37]. It should be noted, however, that ATX, dosed once daily in the present study, may require twice-daily dosing to achieve its maximum beneficial effect [38].
A similar proportion of patients in both treatment groups reported TEAEs; most TEAEs were mild or moderate in severity. The proportions of patients who withdrew from the study as a result of a TEAE were also similar. The most frequently reported TEAEs in the LDX treatment group (decreased appetite, decreased weight, headache, nausea and insomnia) and in patients receiving ATX (headache, nausea, somnolence, decreased appetite and fatigue) are consistent with findings from previous studies [9, 11–13, 28–30]. There was a greater decrease in mean weight in patients receiving LDX than ATX, and more patients receiving LDX than ATX met the outlier criterion for weight loss (≥7 % reduction from baseline). In addition, more patients receiving LDX (n = 4) than ATX (n = 1) moved into the underweight BMI category, all of whom were in the healthy weight (low) category at baseline. These results are consistent with previously reported evidence that psychostimulants are associated with loss of appetite and weight loss. One systematic review reported that children treated with psychostimulants showed a height deficit of approximately 1 cm/year during the first 1–3 years of treatment [39] and clinical guidelines recommend that patients receiving ADHD medication are monitored for weight, height, BMI and appetite every 6 months [40, 41]. High calorific snacks, late evening meals, dosing after meals, drug holidays, or switching to a different class or formulation of medication may be beneficial in some patients [41].
The mean increases in blood pressure and pulse rate observed in both treatment arms in this study were relatively modest. It is recognized, however, that some patients receiving ADHD medications may experience blood pressure and pulse rate above the 95th percentile, and clinical guidelines recommend that patients are assessed for heart disease or symptoms suggesting significant cardiovascular disease, and family history of sudden unexpected death before commencing treatment [40, 41]. Once on medication, patients’ blood pressure and heart rate should be monitored at least every 6 months and, if measurements are above the 95th percentile, it is recommended that patients have a dose-reduction or drug holiday, or are referred to a cardiologist [41]. In the present study, a higher proportion of patients receiving ATX than LDX met the outlier criterion for high pulse rate. With the exception of weight and pulse rate, changes in mean vital sign and ECG parameters, and in the frequency of outliers and PCI observations, were similar between treatment groups.
The strengths of this study include its head-to-head, randomized, double-blind, parallel-group, dose-optimized design, and the large number of patients enrolled from multiple countries. These results are particularly relevant to the recent approval of LDX in Europe for the treatment of children and adolescents whose previous MPH treatment is considered clinically inadequate. However, it is unclear whether this patient population, who met detailed inclusion/exclusion criteria specifically related to prior MPH response, would have favoured a response in one treatment arm over the other. Also, as noted earlier, certain elements of the study design (the 9-week duration and once-daily dosing regimen) may not have elicited the maximum potential treatment benefit of ATX [36, 38].
5 Conclusions
A clinically relevant difference in efficacy was observed between LDX and ATX, with LDX associated with a significantly faster, and more robust, treatment response in children and adolescents with ADHD of at least moderate severity and a previous inadequate response to MPH therapy. Both treatments displayed safety profiles consistent with findings from previous clinical trials. These findings will aid clinicians when developing treatment plans for patients who have achieved unsatisfactory improvements on MPH therapy.
References
Polanczyk G, de Lima MS, Horta BL, Biederman J, Rohde LA. The worldwide prevalence of ADHD: a systematic review and metaregression analysis. Am J Psychiatry. 2007;164(6):942–8.
Hodgkins P, Shaw M, McCarthy S, Sallee FR. The pharmacology and clinical outcomes of amphetamines to treat ADHD: does composition matter? CNS Drugs. 2012;26(3):245–68.
American Academy of Pediatrics. ADHD: clinical practice guideline for the diagnosis, evaluation, and treatment of attention-deficit/hyperactivity disorder in children and adolescents. Process of care supplemental appendix. Pediatrics. 2011a;128:SI1-21. http://pediatrics.aappublications.org/content/suppl/2011/10/11/peds.2011-2654.DC1/zpe611117822p.pdf. Accessed 04 April 2013.
National Institute for Health and Clinical Excellence. Diagnosis and management of ADHD in children, young people and adults. National Clinical Practice Guideline Number 72. London, UK; 2009. http://wwwniceorguk/nicemedia/live/12061/42060/42060pdf. Accessed 04 April 2013.
Hammerness P, McCarthy K, Mancuso E, Gendron C, Geller D. Atomoxetine for the treatment of attention-deficit/hyperactivity disorder in children and adolescents: a review. Neuropsychiatr Dis Treat. 2009;5:215–26.
Sallee FR, Eaton K. Guanfacine extended-release for attention-deficit/hyperactivity disorder (ADHD). Expert Opin Pharmacother. 2010;11(15):2549–56. doi:10.1517/14656566.2010.517523.
Croxtall JD. Clonidine extended-release: in attention-deficit hyperactivity disorder. Paediatr Drugs. 2011;13(5):329–36.
Pennick M. Absorption of lisdexamfetamine dimesylate and its enzymatic conversion to d-amphetamine. Neuropsychiatr Dis Treat. 2010;6:317–27.
Adler LA, Goodman DW, Kollins SH, Weisler RH, Krishnan S, Zhang Y, et al. Double-blind, placebo-controlled study of the efficacy and safety of lisdexamfetamine dimesylate in adults with attention-deficit/hyperactivity disorder. J Clin Psychiatry. 2008;69(9):1364–73.
Biederman J, Boellner SW, Childress A, Lopez FA, Krishnan S, Zhang Y. Lisdexamfetamine dimesylate and mixed amphetamine salts extended-release in children with ADHD: a double-blind, placebo-controlled, crossover analog classroom study. Biol Psychiatry. 2007;62(9):970–6.
Biederman J, Krishnan S, Zhang Y, McGough JJ, Findling RL. Efficacy and tolerability of lisdexamfetamine dimesylate (NRP-104) in children with attention-deficit/hyperactivity disorder: a phase III, multicenter, randomized, double-blind, forced-dose, parallel-group study. Clin Ther. 2007;29(3):450–63.
Coghill D, Banaschewski T, Lecendreux M, Soutullo C, Johnson M, Zuddas A, et al. European, randomized, phase 3 study of lisdexamfetamine dimesylate in children and adolescents with attention-deficit/hyperactivity disorder. Eur Neuropsychopharmacol. 2013;. doi:10.1016/j.euroneuro.2012.11.012.
Findling RL, Childress AC, Cutler AJ, Gasior M, Hamdani M, Ferreira-Cornwell MC, et al. Efficacy and safety of lisdexamfetamine dimesylate in adolescents with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2011;50(4):395–405.
Wigal SB, Kollins SH, Childress AC, Squires L. A 13-hour laboratory school study of lisdexamfetamine dimesylate in school-aged children with attention-deficit/hyperactivity disorder. Child Adolesc Psychiatry Ment Health. 2009;3(1):17.
Wigal T, Brams M, Gasior M, Gao J, Squires L, Giblin J. Randomized, double-blind, placebo-controlled, crossover study of the efficacy and safety of lisdexamfetamine dimesylate in adults with attention-deficit/hyperactivity disorder: novel findings using a simulated adult workplace environment design. Behav Brain Funct. 2010;6:34.
ICH harmonised tripartite guideline: guideline for good clinical practice E6(R1). Geneva: International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, 1996.
American Psychiatric Association. Attention-deficit and disruptive behavior disorders. Diagnostic and statistical manual of mental disorders, 4th ed. Text revision. DSM-IV-TR™ Washington, DC: American Psychiatric Association; 2000. p. 85–93.
Guy W. ECDEU assessment manual for psychopharmacology. Rockville, MD: US Department of Health, Education, and Welfare, Public Health Service, Alcohol, Drug Abuse, and Mental Health Administration, NIMH, Psychopharmacology Research Branch Division of Extramural Research Programs; 1976. p. 218–22.
DuPaul GJ, Power T, Anastopoulos A, Reid R. The ADHD Rating Scale-IV, checklist, norms, and clinical interpretation. New York: Guildford Press; 1998.
Mozzicato P. MedDRA: an overview of the medical dictionary for regulatory activities. Pharm Med. 2009;23(2):11.
Centers for Disease Control and Prevention. http://www.cdc.gov/growthcharts/percentile_data_files.htm. Accessed 04 April 2013.
Hughes CW, Rintelmann J, Emslie GJ, Lopez M, MacCabe N. A revised anchored version of the BPRS-C for childhood psychiatric disorders. J Child Adolesc Psychopharmacol. 2001;11(1):77–93.
Lingjaerde O, Ahlfors UG, Bech P, Dencker SJ, Elgen K. The UKU Side Effect Rating Scale. A new comprehensive rating scale for psychotropic drugs and a cross-sectional study of side effects in neuroleptic-treated patients. Acta Psychiatr Scand Suppl. 1987;334:1–100.
Posner K, Oquendo MA, Gould M, Stanley B, Davies M. Columbia Classification Algorithm of Suicide Assessment (C-CASA): classification of suicidal events in the FDA’s pediatric suicidal risk analysis of antidepressants. Am J Psychiatry. 2007;164(7):1035–43.
Lan KK, Rosenberger WF, Lachin JM. Sequential monitoring of survival data with the Wilcoxon statistic. Biometrics. 1995;51(3):1175–83.
Prentice RL. Linear rank tests with right censored data. Biometrika. 1978;65(1):12.
Cohen J. A power primer. Psychol Bull. 1992;112(1):155–9.
Michelson D, Adler L, Spencer T, Reimherr FW, West SA, Allen AJ, et al. Atomoxetine in adults with ADHD: two randomized, placebo-controlled studies. Biol Psychiatry. 2003;53(2):112–20.
Michelson D, Allen AJ, Busner J, Casat C, Dunn D, Kratochvil C, et al. Once-daily atomoxetine treatment for children and adolescents with attention deficit hyperactivity disorder: a randomized, placebo-controlled study. Am J Psychiatry. 2002;159(11):1896–901.
Michelson D, Faries D, Wernicke J, Kelsey D, Kendrick K, Sallee FR et al. Atomoxetine in the treatment of children and adolescents with attention-deficit/hyperactivity disorder: a randomized, placebo-controlled, dose-response study. Pediatrics. 2001;108(5):E83.
Jain R, Babcock T, Burtea T, Dirks B, Adeyi B, Scheckner B, et al. Efficacy of lisdexamfetamine dimesylate in children with attention-deficit/hyperactivity disorder previously treated with methylphenidate: a post hoc analysis. Child Adolesc Psychiatry Ment Health. 2011;5(1):35.
Hodgkins P, Shaw M, Coghill D, Hechtman L. Amfetamine and methylphenidate medications for attention-deficit/hyperactivity disorder: complementary treatment options. Eur Child Adolesc Psychiatry. 2012;21(9):477–92.
Wehmeier PM, Dittmann RW, Banaschewski T, Schacht A. Does stimulant pretreatment modify atomoxetine effects on core symptoms of ADHD in children assessed by quantitative measurement technology? J Atten Disord. 2012. doi:108705471244518410.1177/1087054712445184.
Newcorn JH, Sutton VK, Weiss MD, Sumner CR. Clinical responses to atomoxetine in attention-deficit/hyperactivity disorder: the Integrated Data Exploratory Analysis (IDEA) study. J Am Acad Child Adolesc Psychiatry. 2009;48(5):511–8.
Newcorn JH, Kratochvil CJ, Allen AJ, Casat CD, Ruff DD, Moore RJ, et al. Atomoxetine and osmotically released methylphenidate for the treatment of attention deficit hyperactivity disorder: acute comparison and differential response. Am J Psychiatry. 2008;165(6):721–30.
Wilens TE, Newcorn JH, Kratochvil CJ, Gao H, Thomason CK, Rogers AK, et al. Long-term atomoxetine treatment in adolescents with attention-deficit/hyperactivity disorder. J Pediatr. 2006;149(1):112–9.
Faraone SV. Using meta-analysis to compare the efficacy of medications for attention-deficit/hyperactivity disorder in youths. P T. 2009;34(12):678–94.
Hanwella R, Senanayake M, de Silva V. Comparative efficacy and acceptability of methylphenidate and atomoxetine in treatment of attention deficit hyperactivity disorder in children and adolescents: a meta-analysis. BMC Psychiatry. 2011;11:176.
Poulton A. Growth on stimulant medication; clarifying the confusion: a review. Arch Dis Child. 2005;90(8):801–6.
Practice parameter for the assessment and treatment of children and adolescents with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2007;46(7):894–921.
Cortese S, Holtmann M, Banaschewski T, Buitelaar J, Coghill D, Danckaerts M, et al. Practitioner review: current best practice in the management of adverse events during treatment with ADHD medications in children and adolescents. J Child Psychol Psychiatry. 2013;54(3):227–46.
Goodman DW, Faraone SV, Adler LA, Dirks B, Hamdani M, Weisler R. Interpreting ADHD rating scale scores: linking ADHD rating scale scores and CGI levels in two randomized controlled trials of lisdexamfetamine dimesylate in ADHD. Primary Psychiatry. 2010;17(3):9.
Acknowledgements
This study was supported by funding from Shire. The authors thank the patients and their parents, and the investigators who took part in the study.
E. Cardo, D. Coghill, R.W. Dittmann and P. Nagy were principal investigators in this clinical study. C.S. Anderson, B. Caballero, R. Civil, N. Higgins, P. Hodgkins and A. Lyne contributed to the study design. R. Bloomfield was responsible for the statistical analysis. All authors were involved in discussion and interpretation of the data, critically revised the article and approved the manuscript before submission. Dr. E. Southam and Dr. T. Gristwood of Oxford PharmaGenesis™ Ltd provided editorial assistance, collated the comments of the authors and edited the manuscript for submission.
C.S. Anderson, R. Bloomfield, B. Caballero, R. Civil, N. Higgins, P. Hodgkins and A. Lyne are employees of Shire and own stock/stock options. The following authors have received compensation for serving as consultants or speakers, or they or the institutions they work for have received research support or royalties from the companies or organizations indicated: E. Cardo (Eli Lilly, Health Spanish Ministry Research Fund, Ministry of Education Grant Research, Shire, UCB); D. Coghill (Flynn Pharma, Janssen-Cilag, Lilly, Medice, Novartis, Otsuka, Oxford University Press, Pfizer, Schering-Plough, Shire, UCB, Vifor Pharma); R.W. Dittmann (Ferring, Janssen-Cilag, Lilly, Otsuka, Shire, the German Research Foundation [DFG], the German Ministry of Education and Research [BMBF], the Ministry of Health/the German Regulatory Body [BfArM], the European Union [EU FP7 program], the US National Institute of Mental Health NIMH, and he is a former employee and stockholder of E. Lilly and Co.); P. Nagy (Tourette Syndrome Association of USA, Hungarian Ministry of Education, National Development Agency of Hungary, Otsuka, Shire).
Author information
Authors and Affiliations
Corresponding author
Additional information
ClinicalTrials.gov NCT01106430.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Dittmann, R.W., Cardo, E., Nagy, P. et al. Efficacy and Safety of Lisdexamfetamine Dimesylate and Atomoxetine in the Treatment of Attention-Deficit/Hyperactivity Disorder: a Head-to-Head, Randomized, Double-Blind, Phase IIIb Study. CNS Drugs 27, 1081–1092 (2013). https://doi.org/10.1007/s40263-013-0104-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-013-0104-8