
Abstract
Background and Objective
Difelikefalin, a selective kappa-opioid receptor agonist, is the first approved treatment for moderate-to-severe pruritus in patients with end-stage renal disease (ESRD) on hemodialysis (HD) in the USA and Europe. The purpose of this open-label study was to investigate the pharmacokinetics and disposition of [14C]difelikefalin following a single intravenous dose in subjects with normal renal function and subjects on HD.
Methods
Twelve adult males (n = 6 healthy subjects; n = 6 subjects on HD) received single intravenous doses of [14C]difelikefalin containing 100 µCi (total doses of 1.7–3.0 μg/kg difelikefalin). Blood, urine, feces, and dialysate samples (when applicable) were collected after dosing.
Results
The median time to maximum concentration was similar for HD and healthy subjects, occurring at 5 min post-dose. The mean area under the concentration–time curve (AUC) was approximately 11-fold higher in HD versus healthy subjects; mean plasma half-life was 38.0 h and 2.6 h, respectively. In healthy subjects, 80.5% of the dose was recovered in urine, and 11.3% was recovered in feces. In subjects on HD, 58.8% of the dose was recovered in feces, and 19.5% was recovered in dialysate [for subjects on HD with residual kidney function (n = 3), 11.2% was recovered in urine]. Based on plasma AUClast, parent [14C]difelikefalin was the most abundant analyte in systemic circulation (> 99% of total exposure) for both cohorts. Metabolite profiles in urine and feces suggested minimal metabolism of the parent compound.
Conclusion
In subjects on HD, difelikefalin total exposure was higher and plasma half-life was longer compared with subjects with intact renal function. Metabolism was low in both healthy subjects and subjects on HD, with unchanged drug representing > 99% of systemic circulation; however, the route of excretion was primarily into urine versus feces in healthy subjects, and feces versus dialysate in subjects on HD.
Registration
ClinicalTrials.gov NCT03947970.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
This study reports key differences in the pharmacokinetics of difelikefalin in subjects on HD relative to healthy subjects, including higher total exposure and longer plasma half-life. |
In healthy subjects, difelikefalin was excreted primarily into urine with metabolites accounting for < 1% of the total dose. In subjects on HD, elimination was primarily through feces and dialysate with a series of low-abundance metabolites identified in feces, ranging from 0.2 to 2.4% of the total dose. |
In both healthy subjects and subjects on HD, difelikefalin was the predominant component in plasma, representing > 99% of circulating radioactivity. |
1 Introduction
Difelikefalin [International Union of Pure and Applied Chemistry name: 4-amino-1-[(2R)-6-amino-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid; molecular weight (free base): 679.4 g/mol (monoisotopic mass), 679.8 g/mol (average); calculated pKa values: 2.4, 7.3, 8.9, and 10.2 (SYBYL, Tripos, Inc., St. Louis, MO)], a selective kappa-opioid receptor (KOR) agonist [1], demonstrated significant reductions in itch intensity versus placebo in adult subjects with moderate-to-severe pruritus on hemodialysis (HD) [2, 3]. In 2021, intravenous (IV) difelikefalin became the first agent approved for the treatment of moderate-to-severe pruritus associated with chronic kidney disease (CKD) in adults on HD [4].
Difelikefalin is a potent, selective KOR agonist with no activity at other receptors, ion channels, or transporters [1]. The unique peptidic structure of difelikefalin differs from other small-molecule KOR agonists developed to date, which mostly are central nervous system (CNS)-active. Difelikefalin, a small hydrophilic d-amino acid peptide [1], was designed to limit membrane permeability by passive diffusion, thereby limiting CNS access. Because difelikefalin does not activate receptors other than KORs mainly located on peripheral sensory neurons and immune cells, it has a more favorable tolerability profile than other opioid agonists, including CNS-active KOR agonists [2, 5, 6]. Difelikefalin has shown no abuse potential, no physical dependence, and no respiratory depression effects; it is not a controlled substance per the Controlled Substances Act in the USA [2, 7, 8].
Pruritus is a chronic, highly burdensome condition in patients with CKD that adversely affects sleep, mood, and ability to socialize [9, 10]. Patients with CKD-associated pruritus (CKD-aP) frequently exhibit considerable mechanical skin damage because of continuous scratching, with excoriations, superimposed infections, and chronic lesions of prurigo nodularis or skin lichenification [11]. Mechanical skin damage can become an additional source of itching. Consequently, patients with CKD-aP are at increased risk of related morbidities associated with infection, e.g., cellulitis, sepsis, bacteremia, cutaneous infections, and infections of the dialysis access [12, 13], and are at higher mortality risk (> 15%) [14, 15]. Large multinational studies (Dialysis Outcomes and Practice Patterns) and studies based in the USA have demonstrated that approximately 20–40% of patients on HD have moderate-to-severe pruritus [15, 16].
The pathophysiology of moderate-to-severe pruritus in patients on HD is not well understood; it is likely that it is multifactorial and that an immune system dysfunction (including elevated proinflammatory activity) and imbalance in the endogenous opioid system (with overexpression of mu-opioid receptors in dermal cells and lymphocytes and concomitant downregulation of KORs) are involved [11, 17,18,19]. Difelikefalin demonstrated antipruritic and anti-inflammatory properties in nonclinical studies [1], and reduced itch intensity in clinical trials of adult subjects with moderate-to-severe pruritus on HD [2, 3].
Here we report results from a phase 1, open-label study assessing the in vivo metabolite profiling and pharmacokinetics (PK) of radiolabeled [14C]difelikefalin administered as a single IV bolus in both healthy subjects and subjects on HD. This mass balance study was conducted to provide quantitative information on the absorption, metabolism, and excretion (AME) of difelikefalin to determine the need for nonclinical safety or drug–drug interaction assessments.
Studies conducted in human hepatic microsomes or in vitro hepatocytes determined that difelikefalin is not metabolized by cytochrome P450 (CYP) enzymes CYP1A2, CYP2C19, CYP2C8, CYP2C9, CYP2D6, or CYP3A [4]. However, little is known about the PK of difelikefalin, and this study is the first to publish data examining the clinical PK of difelikefalin in subjects with normal renal function and subjects on HD who have moderate-to-severe pruritus.
2 Methods
2.1 Study Drug and Formulation
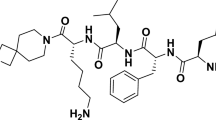
[14C]difelikefalin (14C(U)-D-Phe-D-Phe-D-Leu-D-Lys-[ɣ(4-N-piperidinyl) amino carboxylic acid) acetate salt (Fig. 1), with specific activity 324.4 mCi/mmol, 100 mCi/mL, and 97.4% radiochemical purity by reverse-phase high performance liquid chromatography (HPLC), as a solution in 0.9% saline was synthesized by Polypeptide Laboratories (San Diego, CA) and supplied in vials containing 3 mL each, with one vial given per subject. Dose preparation was conducted under sterile conditions on the day of dosing, sterile filtered, and tested for endotoxins before dosing.

Structure of [14C]difelikefalin. The asterisks denote the uniform C-14 radiolabel incorporated into the phenyl ring of the N-terminal d-phenylalanine
2.2 Patient and Subject Selection
All subjects were nonsmokers, were ≥ 18 years of age, and voluntarily provided written informed consent. All subjects were willing and able to stay in the study center until discharge criteria were met (estimated to be approximately 7 days for healthy subjects and approximately 2 weeks for subjects on HD).
Additional inclusion criteria for healthy subjects included a body mass index (BMI) within the range of 18‒30 kg/m2, inclusive, and body weight ≥ 75 kg. Healthy subjects were in good health as determined by past medical history, physical examination, electrocardiogram, vital sign assessments, and clinical laboratory tests at screening.
Eligible subjects with end-stage renal disease (ESRD) underwent HD 3 times/week for ≥ 3 months prior to screening and demonstrated adequate dialysis measurements (at least 2 Kt/V measurements ≥ 1.2 or 2 urea reduction ratio [URR] measurements ≥ 65%) during the 3 months prior to screening. In addition, subjects had prescription dry body weight ≥ 75 kg and ≤ 135 kg at screening. Subjects taking concomitant medications were on stable doses for at least 14 days prior to dosing, and the dosing regimen had to remain stable for the duration of the study. For a minimum of 1 month prior to dosing, subjects on HD had stable controlled blood pressure that, in the investigator’s opinion, would not interfere with study conduct.
Subjects were excluded from study participation if they had a concomitant disease or any medical condition that, in the opinion of the investigator, could pose undue risk to the participant, could impede completion of the study procedures, or could compromise the validity of the study measurements, including, but not limited to, known or suspected history of alcohol, narcotic, or other drug abuse, or substance dependence within 12 months prior to screening; significant systolic or diastolic heart failure (e.g., New York Heart Association Class IV congestive heart failure); or severe mental illness or cognitive impairment (e.g., dementia). Subjects were also excluded if they had irregular bowel habits (defined as not having bowel movements at least every 2 days); had donated blood or had an acute loss of blood (> 500 mL) during the 3 months prior to study drug administration; or had a history or presence of hypersensitivity or idiosyncratic reaction to opioids.
Further exclusion criteria included positive results for human immunodeficiency virus (HIV), hepatitis B surface antigen, or hepatitis C virus (HCV) antibody at screening (although subjects on HD who tested positive for HCV antibody were allowed to enroll at the discretion of the investigator if their liver function tests were not otherwise clinically significant); previous administration of any [14C]-labeled drug substance within 1 year of study drug administration; or significant (≥ 50 mrems) radiation exposure (such as serial X-rays, computed tomography scans, barium studies, or occupational exposure) within the last 12 months. Healthy subjects were excluded if screening hemoglobin was less than the lower limit of normal. Subjects on HD were excluded if serum alanine aminotransferase or aspartate aminotransferase levels were > 2.5 times the reference upper limit of normal (ULN), or bilirubin was > 4 times the ULN at screening. Please refer to Supplemental Appendix section 2.1 for additional exclusion criteria.
2.3 Clinical Conduct
This was a phase 1 open-label, single-radiolabeled dose, non-randomized study, conducted in compliance with the Declaration of Helsinki, International Council for Harmonisation Good Clinical Practice Guidelines, and all local regulatory requirements.
Subjects received a single dose of study drug, [14C]difelikefalin solution in 0.9% NaCl as a 1 mL IV bolus. For healthy subjects, an IV catheter was inserted at the level of the antecubital fossa or distal to it for study drug administration. Subjects on HD were dosed using their dialysis port after completion of dialysis on the last dialysis day of the week (e.g., Friday for Monday–Wednesday–Friday schedules or Saturday for Tuesday–Thursday–Saturday schedules). The dialysis circuit was disconnected from the dialysis needles prior to dosing; no dosing occurred during rinseback.
The dose of 100 μCi (equivalent to approximately 200 μg of difelikefalin) is considered a standard dose for human AME studies [20, 21], and this level was deemed safe after review of the dosimetry report. The actual body-weight-adjusted [14C]difelikefalin dose contained in the fixed dose of 100 μCi corresponded to a total difelikefalin dose ranging from 1.7 to 3.0 μg/kg across individual participants. Although the recommended dose of difelikefalin in adults with moderate-to-severe pruritus associated with CKD who are on HD is 0.5 μg/kg [4], the higher dose was needed to maximize the ability to detect potential metabolism/degradation products.
All subjects were kept within the study center until (1) concentrations of radioactivity in urine and feces were below quantifiable levels, (2) ≥ 95% of the administered radioactivity was excreted (criterion was set at ≥ 90% for subjects on HD), or (3) sample collections contained ≤ 0.5% of administered radioactivity for 2 consecutive days. Subjects on HD continued to undergo dialysis 3 times per week for 4 h per session until discharge. The dialyzer was either a Fresenius F160 or F180 (Fresenius Medical Care North America, Waltham, MA) with blood flow rates of 400‒500 mL/min and dialysate flow rates of 600 mL/min.
2.4 Sample Collection, Handling, and Storage
For all subjects, samples of blood, urine, and feces were collected at specified times to evaluate the PK, mass balance, and metabolite analysis of [14C]difelikefalin. For subjects on HD, dialysate was collected in hourly intervals during each dialysis session to evaluate mass balance. The timing of sample collection is described in the Supplemental Appendix section 2.3.
2.5 Sample Analysis
Aliquots of plasma, urine, and dialysate samples were measured by pipetting directly into vials, and liquid scintillation fluid was added. Whole blood samples were vortexed, and aliquots were weighed and transferred to combustion cones, dried, and combusted in an oxidizer.
Fecal samples were homogenized with a predetermined solvent; aliquots of the homogenate were transferred to combustion cones, dried, and combusted in an oxidizer.
The concentration of radioactivity in each sample was determined by liquid scintillation counting (LSC) using a Model LS6000 liquid scintillation analyzer (Beckman Coulter, Brea, CA), as described in Supplemental Appendix section 2.4; total radioactivity data were converted to ng- equivalents (ng-eq) or μg-equivalents (μg-eq) for reporting purposes and subsequent calculations.
2.6 Pharmacokinetic Analysis
Data for total radioactivity in plasma, whole blood, urine, feces, and dialysate (for subjects on HD) were analyzed using Phoenix™ WinNonlin® (Version 8.1, Certara, L.P., West Windsor, NJ). The PK population included all participants who received study drug and had sufficient data to calculate the planned PK parameters.
2.6.1 Plasma and Whole Blood
Plasma and whole blood total radioactivity concentrations were analyzed using noncompartmental methods. PK analysis was performed using actual sample times and the actual dose of [14C]difelikefalin for each participant. The following PK parameters were calculated for total radioactivity in plasma and whole blood: concentration at time-zero for IV dosing (C0), maximum observed concentration (Cmax), time to reach Cmax (tmax), area under the concentration–time curve from time-zero to the time of the last quantifiable concentration (AUClast), area under the concentration–time curve extrapolated to infinity (AUCinf), observed terminal rate constant (λz), terminal half-life (t1/2), total systemic clearance (CL), and volume of distribution in the terminal elimination phase (Vz).
2.6.2 Urine and Feces
Urine and feces total radioactivity concentrations were analyzed using nominal collection intervals. The amount of radioactivity excreted in each collection interval was used to derive the following PK parameters: total amount excreted, renal clearance, and feces clearance. The amount of radioactivity excreted was also expressed as a fraction of the administered dose, expressed as a percentage.
2.6.3 Dialysate (Applicable Only to HD Subjects)
Dialysate samples collected at pre-dose (on day 1) and post-dose from subsequent dialysis treatments were reported and summarized. Dialysate sample [14C]difelikefalin equivalents and percentage of dose recovered in dialysate were reported. The PK parameters for dialysate were analogous to those calculated for excreta and included the total amount and the percentage of dose eliminated during dialysis (calculated as the sum of all dialysis treatments).
2.6.4 Mass Balance
Mass balance was assessed by calculating the amount of total radioactivity collected in urine and feces (and dialysate for subjects on HD) relative to the amount of drug administered (radioactive dose). The total recovery relative to dose was expressed as a percentage for each subject reported for each sample collection time interval (by day) as well as the cumulative percentages across the entire sampling duration. Mass balance results were summarized separately for HD and healthy subjects.
2.7 Metabolite Profiling
Samples for metabolite identification in plasma, urine, and feces were prepared by combining an equal volume (plasma), an equal volume fraction of the total volume collected (urine), or an equal weight fraction of the total weight collected (feces) from each subject at a given time point or collection interval. These pooled plasma, urine, and fecal homogenate samples were extracted and analyzed by HPLC. A fraction collector was used to distribute the effluent from the HPLC column into 336 discrete fractions. These discrete fractions were collected in 384-well microplates and analyzed on a scintillation counter to identify radiolabeled species by retention time. Supplemental fractions were then collected at retention times of interest and analyzed by mass spectroscopy (MS) to provide mass-to-charge ratios (m/z) as well as fragmentation patterns for confirmation of molecular structures where possible. Analyses were conducted separately for HD and healthy subjects.
Where possible, difelikefalin and metabolites were quantified by liquid chromatography with tandem mass spectrometry. The relative abundance of difelikefalin and identified metabolites was described for each matrix (plasma, urine, and feces).
3 Results
3.1 Subjects
The characteristics of the six healthy subjects and six subjects on HD who were enrolled and dosed with [14C]difelikefalin are summarized in Table 1. All 12 participants had sufficient data to calculate the planned PK parameters; 2 withdrew consent and discontinued the study early due to personal reasons (1 healthy subject with PK samples collected through 168 h; 1 patient on HD with PK samples collected through 360 h), although with no impact on plasma/blood PK analysis and very little impact on total recovery analysis.
3.2 Pharmacokinetic Analysis
Concentration–time data of total radioactivity in plasma are shown in Fig. 2, and the PK parameters are summarized in Table 2. For total radioactivity in plasma, Cmax and tmax were similar for subjects on HD and healthy subjects; mean Cmax was 25.6 ng-eq/mL and 24.8 ng-eq/mL, respectively, and occurred 5 min post-dose for both groups. Mean AUC was approximately 11-fold higher for subjects on HD compared with healthy subjects; mean AUCinf was 474 h*ng-eq/mL in subjects on HD and 44.3 h*ng-eq/mL in healthy subjects. Mean plasma t1/2 of [14C] equivalents was appreciably longer for subjects on HD (38.0 h) compared with healthy subjects (2.60 h), and mean CL of [14C] equivalents was lower for subjects on HD (0.448 L/h) compared with healthy subjects (4.70 L/h). Mean Vz was slightly higher for subjects on HD (23.4 L) compared with healthy subjects (17.0 L). Vz was relatively low (17.0–23.4 L) and not clinically meaningful in this study, which was expected for a hydrophilic drug with limited membrane permeability that does not partition into body fat.

Mean total radioactivity in plasma after administration of [14C]difelikefalin to healthy volunteers and subjects on hemodialysis
Concentrations of total radioactivity in whole blood were lower but paralleled concentrations in plasma. Based on mean Cmax and AUCinf, total radioactivity in whole blood was approximately 38% (subjects on HD) to 45% (healthy subjects) lower than that in plasma.
3.3 Mass Balance
Percent recovery of total radioactivity is presented in Table 3. In healthy subjects, mean total percent of radioactive dose recovered in urine and feces was 91.8%, with 80.5% of the radioactive dose recovered in urine and 11.3% recovered in feces. The majority of the radioactive dose that was recovered in urine occurred within 24 h of dosing.
In subjects on HD, the mean total percent of radioactive dose recovered in urine, feces, and dialysate was 83.8%, with 11.2% of the radioactive dose recovered in urine, 58.8% recovered in feces, and 19.5% recovered in dialysate. Urinary recovery was based on three subjects, as three of the six subjects produced no urine.
3.4 Metabolite Profiling
Metabolites were numbered based on liquid chromatography (LC) retention time and numbered separately for plasma, urine, and feces. Because of the low masses associated with each metabolite peak, structural assessments were not possible. Therefore, it is unknown if the chromatographic peaks observed in this study are metabolites or degradation products.
3.4.1 Plasma
A representative radiochromatogram in plasma is provided in the Supplemental Appendix (Fig. S1). Concentration–time data are summarized in Table 4 (ng-eq/mL) and Table 5 (relative abundance). Unchanged difelikefalin (parent) was the major component in plasma, and four small metabolite peaks (MP1, MP2, MP3, and MP4) were identified.
Difelikefalin concentrations were higher than metabolite concentrations. In general, only small differences were observed between healthy subjects and subjects on HD. Of the metabolites, MP1 was detected in plasma over a longer time period (0.5–1 h). Other metabolites were detected in only the 0.083- and 0.25-h samples. Difelikefalin was the most abundant analyte in systemic circulation, followed by MP1, MP3, MP2, and MP4; MP2 and MP4 were not detected in subjects on HD. However, the limited number of time points with measurable metabolite concentrations does not allow the relative magnitude of the metabolites (particularly for MP2, MP3, and MP4) to be fully characterized.
Based on AUClast, parent difelikefalin was the most abundant analyte in systemic circulation (> 99% of total exposure) for both healthy subjects and subjects on HD. In healthy subjects, MP1 accounted for 0.48% of the total exposure; AUClast could not be estimated for MP2, MP3, and MP4. In subjects on HD, MP1 accounted for 0.10% and MP3 accounted for 0.02% of the total exposure; AUClast could not be estimated for MP2 and MP4.
3.4.2 Urine
A representative radiochromatogram in urine is provided in the Supplemental Appendix (Fig. S2). Relative abundance of the parent and metabolites is summarized in Table 6. Observed metabolite profiles in urine suggested minimal metabolism of the parent compound. Difelikefalin was the most abundant analyte in urine; traces of two metabolites (MU1 and MU2) were found in subjects on HD, and traces of MU2 and two additional metabolites (MU3 and MU4) were found in healthy subjects. Mass spectrometry characterization of these species was not undertaken due to their low relative abundance. Total percent of dose excreted in urine from the pooled profiling samples (76.8% in healthy subjects, and 12.4% in subjects on HD; Table 6) was comparable to the mean recovery from the mass balance (80.5% in healthy subjects, and 11.2% in subjects on HD; Table 3).
3.4.3 Feces
A representative radiochromatogram in feces is provided in the Supplemental Appendix (Fig. S3). Relative abundance of the parent and metabolites is summarized in Table 7. Observed metabolite profiles in fecal homogenate showed little metabolism of the parent compound in either healthy subjects or subjects on HD. In healthy subjects, almost all of the radioactivity excreted in feces was recovered as unchanged difelikefalin. Metabolites MF4 and MF5 were found in small amounts (< 0.3% LC-radiochromatogram peak area at any given time interval); MS characterization of these species was not undertaken due to their low relative abundance. In subjects on HD, who generally do not produce urine, 84.8% of total radioactivity excreted in feces was recovered as unchanged difelikefalin, and a series of low-abundance metabolites was found (0.187–2.44% of dose).
Total percent of dose excreted in feces from the pooled profiling samples (12.5% in healthy subjects, and 64.9% in HD subjects; Table 7) was comparable to the mean recovery from the mass balance (11.3% in healthy subjects, and 58.8% in subjects on HD; Table 3). Fecal excretion was the dominant pathway for elimination in subjects on HD.
3.5 Safety
There were no serious adverse events (AEs) or AEs leading to participant withdrawal or death. Treatment-emergent AEs (TEAEs) were reported by all six subjects on HD and three of the six healthy subjects. The most frequently reported TEAEs were dizziness (n = 3 subjects on HD; n = 1 healthy subject), headache (n = 4 subjects on HD), paresthesia (n = 2 subjects on HD; n = 1 healthy subject), and nausea (n = 3 subjects on HD). All but one TEAE (moderate constipation) were mild, and the majority (17 of 24 events) were considered by the investigator to be unrelated to study treatment. The most frequently reported TEAEs considered related to treatment were paresthesia (n = 2 subjects on HD; n = 1 healthy subject) and dizziness (n = 1 patient on HD; n = 1 healthy subject) were the most frequently reported treatment-related TEAEs.
4 Discussion and Conclusions
Difelikefalin is a novel, selective KOR agonist approved for the treatment of CKD-aP; there are currently no other approved treatments for this indication in the USA or Europe [2]. In two phase 3 clinical trials in subjects with moderate-to-severe pruritus on HD, treatment with difelikefalin significantly reduced pruritus intensity and improved itch-related quality of life [2, 3]. Consistent with this target treatment population, the current study reports the PK profile, mass balance, and extent of metabolism after a single radiolabeled dose of difelikefalin in both healthy subjects and subjects on HD.
4.1 Pharmacokinetics and Mass Balance
This study demonstrated that the mean plasma Cmax and tmax for difelikefalin were similar in subjects on HD and healthy subjects, occurring 5 min post-dose for both groups. Mean AUC was approximately 11-fold higher for subjects on HD compared with healthy subjects, with longer mean plasma half-life and lower mean total clearance. The observed differences in the difelikefalin PK profile between subjects on HD and healthy subjects in whole blood were analogous to those observed in plasma. The administered radioactive dose was eliminated primarily in urine for healthy subjects and primarily in feces and dialysate for subjects on HD.
4.2 Metabolism
The metabolite profiling analysis indicated only minimal metabolism of this compound, as unchanged difelikefalin represented > 99% of circulating radioactivity in plasma samples from both healthy subjects and subjects on HD. Overall, the results of the metabolic profiling and quantitation reflected the results of the mass balance analysis.
In urine, excretion of unchanged difelikefalin was high in healthy subjects (75.2% of dose) but significantly lower (7.86% of dose) in subjects on HD. Each of the metabolites identified in urine accounted for < 1% of the total dose excreted for both groups.
In feces, unchanged difelikefalin accounted for 11.7% of dose excreted by healthy subjects and 55.1% of dose excreted by subjects on HD. In healthy subjects, conversion of difelikefalin into other species accounted for < 1% of dose, while in subjects on HD, 15.2% of fecal radioactivity was recovered as a series of low-abundance metabolites identified as MF1 through MF9, individually representing 0.187–2.44% of dose. It is possible that the extended in vivo residence time led to a greater number of low-intensity metabolites in subjects on HD, with possible contributions from the action of intestinal bacteria.
4.3 Safety
This single-dose, phase 1 study was conducted in a limited number of healthy subjects and subjects on HD; difelikefalin appeared to have an acceptable safety and tolerability profile with no serious AEs reported. The most common TEAEs were dizziness, headache, paresthesia, and nausea. The majority of TEAEs were reported as mild and considered unrelated to study treatment, similar to safety studies of longer durations. The safety and tolerability profile of difelikefalin was more fully assessed in both 12-week phase 3 clinical trials and a 52-week safety analysis of four phase 3 clinical trials in subjects on HD with moderate-to-severe pruritus [2, 3, 6].
4.4 Limitations
A limitation of this study is the small sample size of subjects with normal renal function and those on HD. While only male subjects were enrolled in this study and were not matched between the two study populations, there are no significant differences in the pharmacokinetics of difelikefalin based on sex or race/ethnicity [4]. This study may have underestimated the amount of drug present in dialysis fluid due to the potential of nonspecific binding to dialysis tubes and membrane. Larger and longer studies have already been conducted that examined the safety of difelikefalin in subjects with moderate-to-severe pruritus on HD [2, 3, 6].
4.5 Conclusions
This phase 1 study defined the PK profile of difelikefalin in both healthy subjects and subjects on HD, determined that elimination was primarily through urine for healthy subjects and primarily in feces and dialysate for subjects on HD, and reported minimal presence of metabolites. Subsequent phase 3 efficacy and safety studies resulted in the first approval of difelikefalin in 2021 as a treatment option for patients on HD with moderate-to-severe CKD-aP, filling an important unmet clinical need in this patient population.
References
Albert-Vartanian A, Boyd MR, Hall AL, Morgado SJ, Nguyen E, Nguyen VP, et al. Will peripherally restricted kappa-opioid receptor agonists (pKORAs) relieve pain with less opioid adverse effects and abuse potential? J Clin Pharm Ther. 2016;41:371–82. https://doi.org/10.1111/jcpt.12404.
Fishbane S, Jamal A, Munera C, Wen W, Menzaghi F. A phase 3 trial of difelikefalin in hemodialysis patients with pruritus. N Engl J Med. 2020;382:222–32. https://doi.org/10.1056/NEJMoa1912770.
Wooldridge TD, Mccafferty K, Schoemig M, Szabolcs Csiky B, Zwiech R, Wen W, et al. Efficacy and safety of difelikefalin for moderate-to-severe CKD–associated pruritus: a global phase 3 study in hemodialysis patients (KALM-2) [abstract FR-OR24]. J Am Soc Nephrol. 2020;31(suppl):22–3.
Korsuva (difelikefalin) [package insert]. Stamford: Cara Therapeutics, Inc.; 2021.
Fishbane S, Mathur V, Germain MJ, Shirazian S, Bhaduri S, Munera C, et al. Randomized controlled trial of difelikefalin for chronic pruritus in hemodialysis patients. Kidney Int Rep. 2020;5:600–10. https://doi.org/10.1016/j.ekir.2020.01.006.
Fishbane S, Wen W, Munera C, Lin R, Bagal S, McCafferty K, et al. Safety and tolerability of difelikefalin for the treatment of moderate to severe pruritus in hemodialysis patients: pooled analysis from the phase 3 clinical trial program. Kidney Med. 2022;4: 100513. https://doi.org/10.1016/j.xkme.2022.100513.
Viscusi ER, Torjman MC, Munera CL, Stauffer JW, Setnik BS, Bagal SN. Effect of difelikefalin, a selective kappa opioid receptor agonist, on respiratory depression: a randomized, double-blind, placebo-controlled trial. Clin Transl Sci. 2021;14(5):1886–93. https://doi.org/10.1111/cts.13042.
Shram MJ, Spencer RH, Qian J, Munera CL, Lewis ME, Henningfield JE, et al. Evaluation of the abuse potential of difelikefalin, a selective kappa-opioid receptor agonist, in recreational polydrug users. Clin Transl Sci. 2022;15:535–47. https://doi.org/10.1111/cts.13173.
Satti MZ, Arshad D, Javed H, Shahroz A, Tahir Z, Ahmed MMH, et al. Uremic pruritus: prevalence and impact on quality of life and depressive symptoms in hemodialysis patients. Cureus. 2019;11: e5178. https://doi.org/10.7759/cureus.5178.
Rehman IU, Lai PSM, Lim SK, Lee LH, Khan TM. Sleep disturbance among Malaysian patients with end-stage renal disease with pruritus. BMC Nephrol. 2019;20:102. https://doi.org/10.1186/s12882-019-1294-1.
Mettang T, Pauli-Magnus C, Alscher DM. Uraemic pruritus—new perspectives and insights from recent trials. Nephrol Dial Transplant. 2002;17:1558–63. https://doi.org/10.1093/ndt/17.9.1558.
Sukul N, Speyer E, Tu C, Bieber BA, Li Y, Lopes AA, et al. Pruritus and patient reported outcomes in non-dialysis CKD. Clin J Am Soc Nephrol. 2019;14:673–81. https://doi.org/10.2215/CJN.09600818.
Ting SW, Fan PC, Lin YS, Lin MS, Lee CC, Kuo G, et al. Uremic pruritus and long-term morbidities in the dialysis population. PLoS ONE. 2020;15:e0241088. https://doi.org/10.1371/journal.pone.0241088.
Narita I, Alchi B, Omori K, Sato F, Ajiro J, Saga D, et al. Etiology and prognostic significance of severe uremic pruritus in chronic hemodialysis patients. Kidney Int. 2006;69:1626–32. https://doi.org/10.1038/sj.ki.5000251.
Pisoni RL, Wikström B, Elder SJ, Akizawa T, Asano Y, Keen ML, et al. Pruritus in haemodialysis patients: International results from the Dialysis Outcomes and Practice Patterns Study (DOPPS). Nephrol Dial Transplant. 2006;21:3495–505. https://doi.org/10.1093/ndt/gfl461.
Rayner HC, Larkina M, Wang M, Graham-Brown M, van der Veer SN, Ecder T, et al. International comparisons of prevalence, awareness, and treatment of pruritus in people on hemodialysis. Clin J Am Soc Nephrol. 2017;12:2000–7. https://doi.org/10.2215/CJN.03280317.
Kimmel M, Alscher DM, Dunst R, Braun N, Machleidt C, Kiefer T, et al. The role of micro-inflammation in the pathogenesis of uraemic pruritus in haemodialysis patients. Nephrol Dial Transplant. 2006;21:749–55. https://doi.org/10.1093/ndt/gfi204.
Tey HL, Yosipovitch G. Targeted treatment of pruritus: a look into the future. Br J Dermatol. 2011;165:5–17. https://doi.org/10.1111/j.1365-2133.2011.10217.x.
Phan NQ, Lotts T, Antal A, Bernhard JD, Stander S. Systemic kappa opioid receptor agonists in the treatment of chronic pruritus: a literature review. Acta Derm Venereol. 2012;92:555–60. https://doi.org/10.2340/00015555-1353.
Beumer JH, Beijnen JH, Schellens JH. Mass balance studies, with a focus on anticancer drugs. Clin Pharmacokinet. 2006;45:33–58. https://doi.org/10.2340/00015555-1353.
McEwen A. The human ADME study. In: Hock FJ, Gralinski MR, editors. Drug discovery and evaluation: methods in clinical pharmacology. Cham: Springer International Publishing; 2017. p. 1–34.
Acknowledgments
The authors thank the study investigators and study participants. We also gratefully acknowledge Amy Shaberman, PhD (Peloton Advantage, LLC, an OPEN Health company, Parsippany, NJ), for medical writing and editorial support, which was funded by Cara Therapeutics, under the direction of the authors.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Cara Therapeutics sponsored this study.
Conflict of interest
JS: worldwide clinical trials—employment. PN: Cara Therapeutics—consultant. RS, SO, and FM: Cara Therapeutics—employment. SB: Cara Therapeutics—consultant.
Availability of data and material
Please contact Cara Therapeutics for data inquiries.
Code availability
Not applicable.
Ethics approval
This study was conducted in compliance with the ethical principles originating in or derived from the Declaration of Helsinki and in compliance with all International Council for Harmonization Good Clinical Practice Guidelines, and all local regulatory requirements were followed.
Consent to participate/consent for publication
All subjects provided written informed consent prior to any baseline study-specific evaluations, per documentation approved by a qualified institutional review board.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Stark, J.G., Noonan, P.K., Spencer, R.H. et al. Pharmacokinetics, Metabolism, and Excretion of Intravenous [14C]Difelikefalin in Healthy Subjects and Subjects on Hemodialysis. Clin Pharmacokinet 62, 1231–1241 (2023). https://doi.org/10.1007/s40262-023-01262-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-023-01262-2