
Abstract
Background and Objective
Spinal muscular atrophy (SMA) is a progressive neuromuscular disease caused by insufficient levels of survival motor neuron (SMN) protein. Risdiplam (EvrysdiTM) increases SMN protein and is approved for the treatment of SMA. Risdiplam has high oral bioavailability and is primarily eliminated through hepatic metabolism by flavin-containing monooxygenase3 (FMO3) and cytochrome P450 (CYP) 3A, by 75% and 20%, respectively. While the FMO3 ontogeny is critical input data for the prediction of risdiplam pharmacokinetics (PK) in children, it was mostly studied in vitro, and robust in vivo FMO3 ontogeny is currently lacking. We derived in vivo FMO3 ontogeny by mechanistic population PK modelling of risdiplam and investigated its impact on drug-drug interactions in children.
Methods
Population and physiologically based PK (PPK and PBPK) modelling conducted during the development of risdiplam were integrated into a mechanistic PPK (Mech-PPK) model to estimate in vivo FMO3 ontogeny. A total of 10,205 risdiplam plasma concentration-time data from 525 subjects aged 2 months–61 years were included. Six different structural models were examined to describe the in vivo FMO3 ontogeny. Impact of the newly estimated FMO3 ontogeny on predictions of drug–drug interaction (DDI) in children was investigated by simulations for dual CYP3A-FMO3 substrates including risdiplam and theoretical substrates covering a range of metabolic fractions (fm) of CYP3A and FMO3 (fmCYP3A:fmFMO3 = 10%:90%, 50%:50%, 90%:10%).
Results
All six models consistently predicted higher FMO3 expression/activity in children, reaching a maximum at the age of 2 years with an approximately threefold difference compared with adults. Different trajectories of FMO3 ontogeny in infants < 4 months of age were predicted by the six models, likely due to limited observations for this age range. Use of this in vivo FMO3 ontogeny function improved prediction of risdiplam PK in children compared to in vitro FMO3 ontogeny functions. The simulations of theoretical dual CYP3A-FMO3 substrates predicted comparable or decreased CYP3A-victim DDI propensity in children compared to adults across the range of fm values. Refinement of FMO3 ontogeny in the risdiplam model had no impact on the previously predicted low CYP3A-victim or -perpetrator DDI risk of risdiplam in children.
Conclusion
Mech-PPK modelling successfully estimated in vivo FMO3 ontogeny from risdiplam data collected from 525 subjects aged 2 months–61 years. To our knowledge, this is the first investigation of in vivo FMO3 ontogeny by population approach using comprehensive data covering a wide age range. Derivation of a robust in vivo FMO3 ontogeny function has significant implications on the prospective prediction of PK and DDI in children for other FMO3 substrates in the future, as illustrated in the current study for FMO3 and/or dual CYP3A-FMO3 substrates.
Clinical Trial Registry Numbers
NCT02633709, NCT03032172, NCT02908685, NCT02913482, NCT03988907.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Previous investigations of FMO3 ontogeny were mostly performed in vitro. Studies in vivo in children are lacking although such data are indispensable for the prediction of paediatric pharmacokinetic and drug–drug interactions (DDIs). |
The in vivo FMO3 ontogeny was estimated using risdiplam data from 525 subjects aged 2 months–61 years by mechanistic population pharmacokinetic (Mech-PPK) modelling. The estimated FMO3 ontogeny suggested (1) higher expression/activity in children with a maximum of threefold difference to adults at 2 years of age; and (2) comparable or lower CYP3A-victim DDI propensity for dual CYP3A-FMO3 substrates in children. |
To our knowledge, this is the first investigation of in vivo FMO3 ontogeny by a middle-out approach using a hybrid of PBPK (bottom-up) and PPK (top-down) modelling. The proposed FMO3 model is recommended for children ≥ 4 months of age. |
1 Introduction
Spinal muscular atrophy (SMA) is an autosomal recessive neuromuscular disease caused by insufficient levels of survival motor neuron (SMN) protein due to the absence of a functional SMN1 gene [1]. The disease manifests with progressive proximal muscle weakness and paralysis and is classified into types 1, 2, 3 and 4, with the age of onset of symptoms being 0–6 months, 7–18 months, > 18 months and 20–30 years, respectively [2]. Risdiplam (EvrysdiTM) modifies splicing of SMN2 pre-mRNA, thereby increasing SMN protein [3]. Risdiplam is approved for the treatment of SMA [4, 5] with doses of 0.2 mg/kg, 0.25 mg/kg and 5 mg for patients aged 2 months to < 2 years, ≥ 2 years and weighing < 20 kg, and ≥ 2 years and weighing ≥ 20 kg [6], respectively. Recently, 0.15 mg/kg of risdiplam has been approved by the US FDA for SMA patients < 2 months of age [7].
Risdiplam has high oral bioavailability (F) following complete absorption, negligible intestinal extraction (< 1%) and low hepatic extraction (EH approximately 5%), as indicated by the human mass-balance and drug–drug interaction (DDI) studies [8, 9]. Risdiplam distributes into peripheral and central tissues [10] with a moderate volume of distribution (Vss = 4.2 L/kg). It is eliminated through hepatic metabolism by flavin-containing mono-oxygenase 3 (FMO3) and cytochrome P450 (CYP) 3A by 75% and 20%, respectively [8, 9], and through renal excretion (5%) [8]. In vitro studies with kidney and intestinal microsomes suggested negligible extrahepatic metabolism [8]. Since FMO3 metabolism is the major elimination route, its ontogeny is a critical physiological input parameter to predict risdiplam clearance (CL) in children. However, there are only a few reports on FMO3 ontogeny, which are mostly based on in vitro investigations [11,12,13]. FMO3 is highly expressed in the adult liver, and catalyses the oxidation of a variety of substrates such as trimethylamine, catecholamines [14], nicotine [15], ranitidine [16] and tamoxifen [17]. Koukouritaki et al. reported an age-dependent increase in FMO3 abundance from neonates to adolescents and adults by approximately 20-fold [11]. A similar age-dependent monotonic increase in FMO3 activity was reported by Xu et al., who quantified FMO3 protein expression in 455 paediatric and adult human liver samples (donors aged 0–87 years) by a quantitative proteomic method [13]. The authors reported an age-dependent 2.2-fold increase in FMO3 expression between neonates and adults and described it using a sigmoidal model [13]. Shimizu et al. [12] evaluated FMO3 expression and trimethylamine N-oxygenation in the liver microsomes (n = 9, aged 13 days–7 years) and by phenotyping in urine (77 children). While the phenotype data demonstrated a monotonic increase in FMO3 activity with age, FMO3 expression and trimethylamine N-oxygenation activity were higher in children than adults [12]. The verification of these reported FMO3 ontogeny with in vivo data are currently limited due to a lack of sensitive FMO3 substrates among drugs used in children.
A physiologically based pharmacokinetic (PBPK) model of risdiplam using these in vitro FMO3 ontogeny models [11, 13] significantly underpredicted apparent clearance (CL/F) in children aged 2 months–18 years [18]. In contrast, application of the ontogeny function by Upreti and Wahlstrom [19] (‘Upreti function’), which describes approximately twofold higher CYP3A expression/activity in children compared with adults based on in vivo data analysis, for both CYP3A and FMO3, adequately predicted the risdiplam CL/F [18]. These findings suggest higher in vivo FMO3 activity in children than predicted by the in vitro FMO3 ontogeny models, warranting a detailed investigation of in vivo FMO3 ontogeny.
Furthermore, the accurate FMO3 ontogeny is an important determinant of victim DDI sensitivity of dual CYP3A-FMO3 substrates such as risdiplam. Although FMO3-mediated DDIs are considered to be clinically rare [20], there is a potential CYP3A-victim DDI risk of concern since numerous CYP3A inhibitors and inducers are used in clinical practice [21]. The paediatric PBPK model of risdiplam with Upreti function for CYP3A and FMO3 ontogeny [18] predicts constant fmCYP3A = 20% in children as in adults, reflecting the assumption of parallel development of these enzymes. However, sensitivity to CYP3A modulation in children relative to adults could be different when enzymes show different developmental patterns [22]. There are several hepatic CYP3A ontogeny functions with different maturation patterns [19, 23,24,25], and currently there is no consensus on their selection [26]. In our previous study [18], different CYP3A ontogeny functions [19, 25, 27] were investigated for extrapolation of the time-dependent inhibition (TDI) of CYP3A of risdiplam from healthy adults to children, accounting for this uncertainty of the hepatic CYP3A ontogeny. In adults, midazolam maximum concentration (Cmax) and area under the concentration-time curve (AUC) were increased by 16 and 11%, respectively, and low CYP3A-TDI propensity was predicted for children across all the CYP3A ontogeny functions examined [18].
The present research investigated in vivo FMO3 ontogeny by mechanistic population pharmacokinetic (Mech-PPK) modelling of risdiplam using the data collected from 525 subjects aged 2 months–61 years. This model is an integration of PPK and PBPK modelling that was iteratively conducted during clinical development (Fig. 1). The impact of the estimated in vivo FMO3 ontogeny on the risdiplam PK, CYP3A-victim DDI and CYP3A-TDI predictions in children was evaluated. Moreover, a simulation study was conducted to demonstrate the implications of the estimated in vivo FMO3 ontogeny on CYP3A-victim DDI propensity of a broad range of dual CYP3A-FMO3 substrates in children. The investigation of in vivo FMO3 ontogeny using the comprehensive data covering a wide age range is also expected to address the current knowledge gap and improve prospective PK and DDI predictions of FMO3 substrates in children.

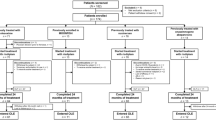
Parallel development of PPK and PBPK models of risdiplam, and integration of the models to develop the Mech-PPK model for investigation of in vivo FMO3 ontogeny. PPK population pharmacokinetic, PBPK physiologically based pharmacokinetic, Mech-PPK mechanistic population pharmacokinetic, ADME absorption, distribution, metabolism, excretion, SMA spinal muscular atrophy, CLint intrinsic clearance, CL/F apparent clearance, PK pharmacokinetics, FMO3 flavin-containing mono-oxygenase 3, CYP cytochrome P450, DDI drug–drug interaction, TDI time-dependent inhibition
2 Methods
The development of PPK, PBPK and Mech-PPK models for risdiplam are summarized in Fig. 1. Plasma samples were collected from healthy individuals or SMA patients who participated in five clinical trials (NCT02633709, NCT03032172, NCT02908685, NCT02913482, NCT03988907; summarized in electronic supplementary material [ESM] Table S1) and the determined risdiplam concentrations (ESM Fig. S1) were used in the modelling. Bioanalytical method, definitions of model evaluation and validation in this report are provided in ESM 1. Initially, PBPK and PPK models were developed with data collected from 130 subjects (26 healthy adults and 104 SMA patients) [ESM Table S2) aged 2 months to 52 years (Fig. 1a–c). Subsequently, the models were evaluated and validated with independent data collected from 395 subjects (35 healthy adults and 360 SMA patients) [ESM Table S2] aged 2 months–61 years (Fig. 1d). The final PPK model parameter estimation and Mech-PPK modelling were performed with all available risdiplam data (total of 130 + 395 = 525 subjects). The median PK observation period was 358 days for SMA patients. The patient demographics, model development and evaluation/validation datasets are summarized in ESM Tables S1 and S2. The Mech-PPK model represents a hybrid of the PPK and PBPK models considering its physiologically based representation of hepatic metabolism while otherwise being empirical. The Mech-PPK model was used to estimate the in vivo FMO3 ontogeny (Fig. 1e, f). This ontogeny function was subsequently implemented in the risdiplam PBPK model and used for CYP3A-victim DDI and -TDI assessments in children (Fig. 1g).
2.1 Initial Physiologically Based Pharmacokinetic (PBPK) and Population Pharmacokinetic (PPK) Model Developments for Risdiplam (Fig. 1a–c)
The PBPK model of risdiplam was previously developed and verified with healthy adult data [8]. In brief, it consists of (1) a mechanistic absorption model; (2) volume of distribution predicted by tissue to plasma partition coefficients according to a preclinical distribution study [10]; (3) hepatic CL by FMO3 (75%) and CYP3A (20%); and (4) renal excretion (5%). This model was used for the selection of starting doses in the therapeutic studies of risdiplam (Fig. 1a). Since plasma samples collected from SMA patients were sparse, PK were analysed by non-linear mixed-effects modelling. An initial PPK model of risdiplam was developed on 2492 observations collected from 130 subjects (ESM Table S3). A PK model consisting of a three-transit compartment for absorption connected to a linear disposition model with two compartments was identified as a suitable structural model. Time-varying body weight as a covariate on CL/F, apparent intercompartmental clearance (Q/F), apparent volume of distribution of the central compartment (Vc/F) and apparent volume of distribution of the peripheral compartment (Vp/F) using an allometric model with estimated or fixed coefficients, and time-varying age as a covariate on CL/F and Vc/F with a sigmoidal model were implemented. The estimated parameters, goodness-of-fit (GOF) plots and prediction-corrected visual predictive check (pc-VPC) are provided in ESM Figs. S2 and S3. The post hoc CL/F of the PPK model was approximately 30% lower in the adult SMA patients than age- and body-weight-matched healthy subjects (ESM Fig. S4). Therefore, risdiplam intrinsic CL (CLint) and renal CL in the PBPK model for SMA patients were reduced by 30% (ESM Table S4). The body-weight-normalized CL/F values were higher in paediatric patients than in adult patients (ESM Fig. S5). Since CL/F is an adequate surrogate of the CLint of risdiplam (exclusively metabolized in the liver and low hepatic extraction), this finding suggested that hepatic metabolic activity per gram of liver in children is higher than adults. Use of existing in vitro FMO3 ontogeny functions [11, 13] (ESM Fig. S6) significantly underpredicted risdiplam CL/F in children (ESM Fig. S7a). The Upreti function for the hepatic CYP3A ontogeny [19] (ESM Fig. S6), applied for scaling of both CLint,CYP3A and CLint,FMO3, adequately predicted risdiplam CL/F in children and was retained in the model (ESM Fig. S7b).
2.2 Evaluation of the PPK Model and Validation of the PBPK Model (Fig. 1d)
An additional 7713 risdiplam plasma samples collected from 395 subjects (35 healthy subjects, 360 SMA patients aged 2 months–61 years) [ESM Table S2] were used in the evaluation of the PPK model. Prediction success of the initial PPK model, after the addition of the new data, was examined. These additional data were used for external validation of risdiplam PBPK models for SMA patients. The predicted risdiplam Cmax, AUC from time zero to 24 h (AUC0-24h) and CL/F were compared with the observed Cmax and individually estimated AUC0-24h and CL/F according to the post-hoc parameters of the PPK model, respectively.
2.3 Mechanistic PPK Modelling of Risdiplam to Estimate in vivo Flavin-Containing Mono-Oxygenase 3 (FMO3) Ontogeny (Fig. 1e, f)
A Mech-PPK model for risdiplam incorporated a physiological description of the hepatic metabolism in the PPK model using a perfusion limited/well-stirred liver model. The following assumptions were made based on the PBPK model: (1) the oral F was equivalent to the hepatic availability (FH) due to complete absorption and negligible intestinal extraction [8]; and (2) the composition of adult CL (fmFMO3:fmCYP3A:renal CL = 75%:20%:5%). Drug absorption and distribution remained empirical, as in the PPK model.
The Mech-PPK model was fitted to all available data (n = 525, 10,205 observations) to estimate the in vivo ontogeny of FMO3. Six different structural models, including combinations of sigmoidal Emax, the Gompertz [28], and the Richards functions [29] (Table 1), were examined to describe the higher expression/activity observed in children than adults. The Upreti function [19] was used for the hepatic CYP3A ontogeny. Postnatal age was used in the ontogeny functions since most infants participated in the risdiplam clinical study were born full-term. Age-dependent physiological parameters (liver size, hepatic blood flow) [24], scaling factors (microsomal protein in the liver) [30, 31] and CL equations are summarized in ESM Eqs. S1–11. Parameter estimates, GOF and pc-VPC were examined. A selected in vivo FMO3 ontogeny model was implemented in the paediatric PBPK model of risdiplam, which was evaluated by comparing the predicted Cmax and AUC with the observations.
2.4 Investigation of Cytochrome P450 (CYP) 3A-Mediated DDI Risk of Dual CYP3A-FMO3 Substrates in Children Using the Estimated in vivo FMO3 Ontogeny Function (Fig. 1g)
The CYP3A-victim DDI risk of risdiplam in adults is low (fmCYP3A = 20%) [8, 9]. However, as risdiplam is a dual CYP3A-FMO3 substrate, age-dependent changes in FMO3 and CYP3A ontogeny may alter CYP3A DDI susceptibility in children relative to adults. The paediatric PBPK model, updated with the estimated in vivo FMO3 ontogeny function, was used to predict risdiplam fmCYP3A and to assess CYP3A-victim DDI propensity in children. The updated model was also used to predict CYP3A-TDI on midazolam PK in children and was compared with the previous prediction [18]. In addition, a simulation study with a broader range of theoretical dual CYP3A-FMO3 substrates was conducted to evaluate the impact of the estimated in vivo FMO3 ontogeny function on CYP3A-victim DDI risk. The theoretical dual substrates with adult fmCYP3A:fmFMO3 ratios of 0.1:0.9 (substrate A), 0.5:0.5 (substrate B) and 0.9:0.1 (substrate C) were evaluated by predicting AUC ratios [18] (CYP3A-AUCR, Eq. 1). No involvement of intestinal metabolism and 95% inhibition of the hepatic CYP3A activity were assumed. The CYP3A-AUCR of children was compared with the adults (AUCRchildren/AUCRadults) to evaluate age-dependent sensitivity to CYP3A modulation.
NONMEM version 7.4, with a first-order conditional estimation with interaction (FOCE-I) method, was used for the PPK and Mech-PPK modelling. All graphical analyses were performed using R version 4.0.3 and RStudio version 1.2.1335. PsN (version 4.9.3) was used for model diagnostics. The PBPK modelling (Fig. 1a, c, d) and CYP3A-victim DDI and -TDI predictions were performed using SimCYP versions 18 and 20, respectively.
3 Results
3.1 Evaluation and Validation of the PPK and PBPK Models of Risdiplam (Fig. 1d)
The initial PPK model (ESM Table S3) was fitted to the combined dataset including the additional 395 subjects (total n = 525 including the 130 subjects in the model development dataset, 10,205 observations). Although the parameter estimates (Table 2) differed from the initial PPK model, the structural model remained the same. An additional covariate, a factor for CL/F of healthy adults (n = 61), and a separate residual error term for capillary blood samples (3% of the dataset) were introduced. All parameters were estimated with good precision, and the pc-VPC (Fig. 2a) and GOF plots (ESM Fig. S8) confirmed the ability of the model to predict central tendency and variability. The estimated allometric coefficient of CL/F was decreased from 0.418 to 0.276, further deviated from the theory-based allometric exponent of 0.75 [32]. The allometric coefficient of Vc/F slightly shifted from 1.0 (fixed) to 0.86 and the Age50 of the maturation functions of CL/F and Vc/F remained comparable (ESM Table S3; Table 2). The resulting population estimates of CL/F and Vc/F of the updated PPK model were generally consistent with the initial PPK model in children (ESM Fig. S9), showing the robustness of the model. Consequently, this model was accepted as the final PPK model.

Evaluation/validation of the PPK, Mech-PPK and PBPK models of risdiplam. a pc-VPC of the PPK model of risdiplam for the dataset that contains 525 subjects. Individual observations corrected by the respective prediction are shown with solid circles. Gray areas are 95% prediction intervals of the 2.5th, median and 97.5th percentiles of predictions. Dotted and solid lines show the 2.5th and 97.5th percentiles and the median of the observations, respectively. b Validation of the risdiplam PBPK model by comparing predicted AUC0-24h (gray shade) with the individually estimated AUC0-24h using post- hoc PK parameters of the final PPK model for the 270 paediatric SMA patients aged 2 months–18 years in the model validation dataset (ESM Table S2) who received an approved dose of risdiplam (0.2, 0.25 mg/kg or 5 mg) [6] for more than 25 days. The default demographic model supplied in the SimCYP paediatric module was used. The gray and striped shapes show the 90% prediction interval or 5th–95th percentiles of the observations, respectively. Geometric means of simulated (solid squares) and observed (open circles) values are shown. Geometric means of the simulated risdiplam AUC0-24h are all within 0.8- to 1.2-fold of the observations, except for AUC0-24h of 2–4 years (1.4). c pc-VPC of the Mech-PPK model of risdiplam with the in vivo FMO3 ontogeny Model 6. Individual observations corrected by the respective population prediction are shown with solid circles. Gray areas are 95% prediction intervals of the 2.5th, median and 97.5th percentiles of predictions. Dotted and solid lines show the 2.5th and 97.5th percentiles and the median of the observations, respectively. d Prediction of risdiplam AUC0-24h by the updated paediatric PBPK model after implementation of the estimated in vivo FMO3 ontogeny function was compared with the individually estimated AUC0-24h using post-hoc PK parameters of the final PPK model for 362 paediatric patients with SMA who received an approved dose of risdiplam (0.2, 0.25 mg/kg or 5 mg) [6] for more than 25 days. A customized demographic model for SMA patients [18] was used. The gray and striped shapes show the 90% prediction interval or the 5th–95th percentiles of the observations, respectively. Geometric means of simulated (solid squares) and observed (open circles) values are shown. Geometric means of the simulated risdiplam AUC0-24h were within 0.8- to 1.2-fold of the observations in children aged 2–7 months and > 4 years, and 0.76- to 0.79-fold in children aged 7 months to 4 years. PPK population pharmacokinetic, PBPK physiologically based pharmacokinetic, Mech-PPK mechanistic population pharmacokinetic, pc-VPC prediction-corrected visual predictive check, AUC0-24h area under the concentration-time curve from time zero to 24 h, PK pharmacokinetic, SMA spinal muscular atrophy, FMO3 flavin-containing mono-oxygenase 3
The paediatric PBPK model adequately predicted AUC (Fig. 2b), Cmax and CL/F of the paediatric SMA patients aged 2 months–18 years and the adult SMA patients (ESM Figs. S11–14) in the external validation dataset. Therefore, these PBPK models for SMA patients were considered satisfactorily validated.
3.2 Estimation of In Vivo FMO3 Ontogeny by Mech-PPK Modelling of Risdiplam (Fig. 1e, f)
After the robustness of the PPK and PBPK models was confirmed, the Mech-PPK model was developed and fitted to all available data (n = 525, 10,205 observations) to estimate in vivo FMO3 ontogeny using six different structural models (Table 1). The predicted FMO3 expression/activity was higher in children, reaching a maximum at 2 years of age with an approximately threefold difference compared with adults (Fig. 3). All six model predictions were consistent from the age of 4 months, whereas divergent predictions were noted for younger infants. Parameter estimation for Model 6, a combination of the Richards function and Emax model, was considered the most reliable (Table 1). Consistency with the bootstrap analysis further supports this model (Table 3, ESM Fig. S15). The pc-VPC (Fig. 2c) and GOF (ESM Fig. S16–18) showed its ability to adequately predict central tendency and variability of risdiplam PK. Therefore, Model 6 (Eq. 2) was selected as a representative in vivo FMO3 ontogeny model.

Estimated in vivo FMO3 ontogeny functions by the mechanistic PPK model of risdiplam. Predictions of in vivo FMO3 ontogeny by the six mechanistic PPK models of risdiplam are shown with solid lines, between 2 months and 50 years of age. The in vivo FMO3 ontogeny functions represent the predicted fraction of adult’s FMO3 activity based on the scaled CLint and accounting for age-dependent liver weight and MPPGL. The hepatic CYP3A4 ontogeny according to Upreti and Wahlstrom [19] (black dotted lines) is shown as a reference. The number of subjects in the analysis dataset between 1–2, 2–3, 3–4 and 4–5 months of age are indicated. Dotted lines in the 1–2 months age group represent extrapolations by these six FMO3 in vivo ontogeny models. FMO3 flavin-containing mono-oxygenase 3, PPK population pharmacokinetic, CLint intrinsic clearance, MPPGL microsomal protein per gram of liver, CYP cytochrome P450
Subsequently, the risdiplam paediatric PBPK model was updated with the estimated in vivo FMO3 ontogeny Model 6. The model predicted risdiplam AUC0-24h (geometric mean) within 0.8- to 1.25-fold of the observations in children aged 2–6 months and > 4 years, and 0.76- to 0.79-fold in children aged 7 months to 4 years (Fig. 2d). The geometric mean of predicted Cmax was 0.6- to 0.75-fold of the observations in children aged 2 months–2 years, and within 0.8- to 1.25-fold in children ≥ 2 years of age (ESM Fig. S20).
3.3 Predictions of CYP3A-Victim Drug–Drug Interaction (DDI) and CYP3A–Time Dependent Inhibition (TDI) of Risdiplam in Children with the Novel in vivo FMO3 Ontogeny Model (Fig. 1g)
The updated PBPK model of risdiplam with the estimated in vivo FMO3 ontogeny predicted comparable or lower fmCYP3A values in children than the previous PBPK model predictions with Upreti function for CYP3A and FMO3 (ESM Fig. S21). The predicted CYP3A-TDI was consistent with the previous work [18], with the predicted midazolam Cmax and AUC ratios constantly < 1.25 in children aged 2 months–18 years (ESM Fig. S22). These DDI predictions for infants 2–4 months of age were repeated with in vivo FMO3 ontogeny Models 2 and 3, representing the lowest and highest FMO3 expression/activity, respectively (Fig. 3, ESM Eqs. S12, S13). Consideration of the uncertainty in FMO3 ontogeny in infants 2–4 months of age had no impact on the predicted fmCYP3A and midazolam AUC/Cmax ratios (ESM Fig. S23). Overall, the predicted CYP3A-victim DDI and -TDI propensity in children aged 2 months–18 years remained low after implementation of the newly estimated in vivo FMO3 ontogeny functions.
3.4 Simulations of CYP3A-Victim DDI Propensity for Different Dual CYP3A-FMO3 Substrates in Children
The impact of the estimated in vivo FMO3 ontogeny function on CYP3A-victim DDI propensity was evaluated for a range of theoretical dual CYP3A-FMO3 substrates in children aged 4 months–18 years, by simulations. The predicted fmCYP3A and resulting CYP3A-AUCR of substrates A–C were generally lower in children < 10 years of age than in adults (Fig. 4a, b), reflecting higher relative FMO3 expression/activity compared with CYP3A in this age range. Regardless of the relative CYP3A-FMO3 contribution, a comparable CYP3A-AUCR with adults (within 10%) was reached at 10 years of age. The difference in predicted CYP3A-AUCR between children <10 years of age and adults depended on the fmCYP3A (Fig. 4b). Simulations highlighted lower CYP3A-victim DDI risk in children, particularly those < 5 years of age, with more than 25% lower CYP3A-AUCR than adults for substrate C (adult fmCYP3A = 90%). For infants aged 2–4 months, predicted fmCYP3A and CYP3A-AUCR were lower than adults among the FMO3 ontogeny Models 2, 3 and 6 (ESM Fig. S24).

CYP3A-victim DDI risk assessments for theoretical dual CYP3A-FMO3 substrates in children using the in vivo FMO3 ontogeny function (Model 6). a Simulated fmCYP3A—age profiles of the theoretical dual CYP3A and FMO3 substrates: fmCYP3A:fmFMO3 of 0.1:0.9 (substrate A—open circle), 0.5:0.5 (substrate B—triangles) and 0.9:0.1 (substrate C—solid squares). The ratios of fmCYP3A:fmFMO3 correspond to values in adults. The hepatic CYP3A ontogeny according to Upreti and Wahlstrom [19], and the estimated in vivo FMO3 ontogeny (Model 6), were used. b The ratios of AUCR (AUCRchildren/AUCRadults) between children and adults for each substrate are shown across the age range. CYP cytochrome P450, DDI drug–drug interaction, FMO3 flavin-containing mono-oxygenase 3, AUCR area under the concentration-time curve ratio
4 Discussion
The Mech-PPK modelling of risdiplam to estimate a novel in vivo FMO3 ontogeny was built on 10,205 risdiplam data points from 525 subjects aged 2 months–61 years with a median PK observation period in SMA patients of 358 days. The model predicts higher FMO3 expression/activity in children compared with adults, reaching a maximum at 2 years of age, with an approximately threefold difference compared with adults. This model substantially improved the prediction of risdiplam PK in children compared with the in vitro FMO3 ontogeny models [11, 13]. To our knowledge, this is the first investigation of in vivo FMO3 ontogeny by a middle-out approach using a hybrid of PBPK (bottom-up) and PPK (top-down) modelling.
The PPK modelling was essential to analyse sparse data collected from a limited number of SMA patients at the beginning of clinical development. It characterized age- and disease-dependent processes in risdiplam PK, which informed the PBPK modelling. The PBPK model supported selection of risdiplam doses for the clinical studies and the DDI predictions for children [18]. After the robustness of these models was confirmed (Fig. 1d), the physiological representations of F and CL, and drug-related assumptions, were implemented in the PPK model to develop the Mech-PPK model. This approach enabled differentiation of CYP3A and FMO3 ontogenies, whereas the maturation function of CL/F in the PPK modelling was indiscriminate of enzyme. The Mech-PPK retained adequate prediction ability of risdiplam PK, as shown by the consistent pc-VPC, GOF (Fig. 2a, c; ESM Figs. S8, S16–18) and the population estimates of CL/F across the age range with the PPK model (ESM Fig. S19). Existing ontogeny functions that describe expression/activity in children exceeding those in adults were provided as discrete fractions [23] or as a continuous model with up to nine parameters [19]. In contrast, the current analysis (Mech-PPK) is equally flexible but represents a more parsimonious model with a continuous function requiring six to seven parameters to describe enzyme ontogeny. Implementation of physiological representations in PPK modelling enabled investigations of age-dependent physiology from infrequently collected data from children. The utility of such an integrated modelling approach was also previously shown in population PBPK modelling of midazolam that investigated the intestinal and hepatic CYP3A ontogeny [33].
A typical allometric exponent of 0.75 for CL reflects the relationship between body size and liver weight (ESM Fig. S25), and therefore it describes CL through hepatic metabolisms across children and adults when they have the same enzyme activity. The Mech-PPK model implemented CL with the estimated liver weight (ESM Eq. S3, ESM Fig. S25) and therefore the allometric exponent was not required. The much lower allometric exponent estimated for risdiplam CL/F of the PPK model (i.e. 0.276) is indicative of higher enzyme activity in children compared with adults. When the allometric scaling is fixed to 0.75, a bias appears in the ETA-CL/F distribution over age (ESM Fig. S26). The correction of this bias would require a maturation function describing higher CL/F between 6 months and 10 years of age relative to adults (ESM Fig. S26), in agreement with the estimated in vivo FMO3 ontogeny by the Mech-PPK modelling reported herein.
To our knowledge, FMO3 ontogeny has mostly been investigated in vitro [11,12,13]. Koukouritaki et al. reported an approximately 20-fold increase in FMO3 abundance from neonates to adolescents [11]. A similar age-dependent trend was reported from the proteomic analysis of the human liver samples [13]. These in vitro data suggested a monotonic increase in FMO3 expression/activity with age and do not exceed adult levels. Application of these in vitro FMO3 ontogeny functions significantly underpredicted risdiplam CL/F in children (ESM Fig. S7). In contrast, Shimizu et al. reported increased FMO3 expression and trimethylamine N-oxygenation in children relative to adults [12]. Although the sample size was small (nine donors), it is noteworthy that the trimethylamine N-oxygenation was the highest at 2 years of age and the difference from adults was three- to fourfold [12], in agreement with the in vivo FMO3 ontogeny models reported herein. Greater metabolic activity in children compared with adults was also reported for multiple CYPs (CYP3A, 1A2, 2C8, 2C9 and 2C19) from investigations of ontogeny using in vivo data [19]. Involvement of multiple factors such as age-dependency in DNA methylation, nuclear receptors, tissue volume and composition, and hormonal changes are suggested for the development of drug-metabolizing enzymes [34]. While hepatocyte nuclear factor-4 binding site and/or CCAAT box are speculated to be responsible for maturation of FMO3 [12, 35], the exact age-dependent mechanism is currently unknown.
The presented research was conducted in SMA patients, like other in vivo ontogeny studies that relied on paediatric patient data [19, 24, 25, 36, 37]. Therefore, the estimated FMO3 ontogeny may not represent other populations. Adult patients with SMA showed approximately 30% lower CL/F than healthy adults, for reasons unknown. The severity of SMA is correlated with age of onset of symptoms [2], with younger patients likely having more severe forms than older patients and this could have confounded the estimation of the ontogeny. There were fewer observations from subjects < 2 years of age (13%) than those > 2 years of age (87%), and data were particularly sparse for patients aged 2–5 months (2%). This might partially explain the minor underprediction of risdiplam exposure in children aged < 4 years of age (Fig. 2d) and higher uncertainty with different predicted trajectories of FMO3 in infants < 4 months of age (Fig. 3, ESM Fig. S15). Genetic polymorphisms of FMO3 with a link to altered function have been reported [12, 13, 38]. FMO3 genotyping was not performed in the clinical studies; however, there was no obvious outlier (ESM Fig. S1) in the database and therefore the influence of FMO3 polymorphism on the ontogeny investigation is considered unlikely. Nevertheless, risdiplam is a suitable substrate to investigate in vivo FMO3 ontogeny because of high fmFMO3 [8], high oral F [8], low hepatic extraction [8] and age-independent plasma protein binding [39]. Moreover, the data used for the investigation of ontogeny were comprehensive and included 382 paediatric patients with SMA ranging from 2 months to 18 years of age, and a median PK observation duration of 439 days and up to 3 years. All six different model structures for in vivo FMO3 ontogeny tested herein predicted consistent FMO3 expression/activity in the age range ≥ 4 months, and therefore confidence in the estimates in this age range is high. The application of Model 6, the current representative in vivo FMO3 ontogeny model, is recommended for PK predictions in children ≥ 4 months of age. Further investigation is needed with more longitudinal data, and validations with other FMO3 substrates using a similar analysis as proposed herein would improve the confidence in this novel FMO3 ontogeny function in the future. Until additional data are generated, it is recommended that FMO3 ontogeny Models 2 and 3 are considered in addition to Model 6 for predictions of children aged < 4 months to account for uncertainty of FMO3 ontogeny in that early age range.
The novel in vivo FMO3 ontogeny function predicts more rapid development of the enzyme and greater FMO3 expression/activity than the Upreti function for the hepatic CYP3A ontogeny (Fig. 3). These differences in development trajectories may lead to altered CYP3A-victim DDI sensitivity of dual CYP3A-FMO3 substrates in children. The analysis of theoretical dual FMO3-CYP3A substrates predicted lower fmCYP3A and CYP3A-AUCR in children aged 4 months–10 years than in adults. CYP3A-victim DDI for substrates with fmCYP3A > 0.8 is especially sensitive to even minor differences in fmCYP3A in the case of potent CYP3A inhibition [40, 41]. Therefore, the FMO3 ontogeny had a more pronounced effect on the predicted CYP3A-AUCR for such substrates, as evidenced by the larger decrease in CYP3A-AUCR in children than in adults for substrate C (adult fmCYP3A = 90%) [Fig. 4b], despite minor FMO3 contribution (10%). The simulations in infants 2–4 months of age with the in vivo FMO3 ontogeny Models 6, 2 and 3 showed comparable or lower CYP3A-AUCR versus adults, even with the most conservative FMO3 ontogeny model (ESM Fig. S24). Overall, lower CYP3A-victim DDI propensity in children ≥2 months of age compared with adults was predicted for dual CYP3A-FMO3 substrates when the newly estimated FMO3 ontogeny was considered. Risdiplam showed same trends, as its predicted fmCYP3A with the newly estimated FMO3 ontogeny model and Upreti function for CYP3A ontogeny was comparable or slightly lower in children compared with adults (ESM Figs. S21, 23b). The use of hepatic CYP3A ontogeny models [23, 25] other than the Upreti function in the investigation of FMO3 ontogeny (e.g., Salem function) [ESM Fig. S6], leads to higher FMO3 expression/activity in children (ESM Fig. S27). A combination of this alternative FMO3 ontogeny function and Salem function for CYP3A ontogeny would predict either a comparable or further decrease in fmCYP3A and CYP3A-AUCR in children, supporting that the use of Upreti function reported herein is more conservative in CYP3A-victim DDI assessment. As a potential perpetrator, risdiplam CYP3A-TDI is predominantly occurring in the intestine [18], and therefore change in the hepatic FMO3 ontogeny is not expected to alter the TDI prediction. Indeed, the predicted inhibitory effect on midazolam PK remained low (Cmax and AUC ratios <1.25) [ESM Figs. S22 and 23c, d] after revision of the FMO3 ontogeny models. Therefore, refinement of FMO3 ontogeny in the risdiplam paediatric PBPK model, including Models 2 and 3 for infants 2–4 months of age, had no impact on the previously predicted low CYP3A-victim DDI and -TDI propensity of risdiplam in children aged from 2 months to 18 years.
5 Conclusion
Mech-PPK modelling of risdiplam successfully estimated the in vivo FMO3 ontogeny from risdiplam data collected from 525 subjects aged 2 months–61 years. The estimated in vivo FMO3 ontogeny predicts higher expression/activity in children reaching a maximum at the age of 2 years, with an approximately threefold difference compared with adults. To our knowledge, this is the first estimation of in vivo FMO3 ontogeny by a middle-out approach using a hybrid of PBPK (bottom-up) and PPK (top-down) modelling and comprehensive data covering a wide age range. Since all structural models investigated herein predicted consistent FMO3 ontogeny in the population ≥ 4 months of age, the application of the function is currently recommended from this age to adulthood. The simulation study with the range of theoretical CYP3A-FMO3 substrates implied comparable or lower CYP3A-victim DDI propensity in children compared with adults when the newly estimated in vivo FMO3 ontogeny is considered. Similarly, comparable or lower CYP3A-victim DDI and -TDI propensity were predicted for risdiplam in children aged ≥ 2 months compared with adults, and refinement of FMO3 ontogeny had no impact on the previously predicted low CYP3A-DDI risk. The novel in vivo FMO3 ontogeny addresses the knowledge gap and is envisaged to improve prospective PK and DDI predictions of FMO3 substrates in children.
References
Lefebvre S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80(1):155–65.
D’Amico A, et al. Spinal muscular atrophy. Orphanet J Rare Dis. 2011;6:71.
Ratni H, et al. Discovery of risdiplam, a selective survival of motor neuron-2 ( SMN2) gene splicing modifier for the treatment of spinal muscular atrophy (SMA). J Med Chem. 2018;61(15):6501–17.
European Medicines Agency. Evrysdi (risdiplam). 2021. https://www.ema.europa.eu/en/documents/smop-initial/chmp-summary-positive-opinion-evrysdi_en.pdf. Accessed Feb 2022.
US FDA. FDA Approves Oral Treatment for Spinal Muscular Atrophy. 2020. https://www.fda.gov/news-events/press-announcements/fda-approves-oral-treatment-spinal-muscular-atrophy. Accessed Nov 2022.
US FDA. EVRYSDI™ (risdiplam) for oral solution. Highlights of Prescribing Information. 2020. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/213535Orig1s000lbl.pdf. Accessed Nov 2022.
US FDA. Highlights of prescribing information. EVRYSDI (risdiplam) for oral solution. 2022. https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/213535s003s005lbl.pdf. Accessed Nov 2022.
Fowler S, et al. Addressing today’s absorption, distribution, metabolism, and excretion (ADME) challenges in the translation of in vitro ADME characteristics to humans: a case study of the SMN2 mRNA splicing modifier risdiplam. Drug Metab Dispos. 2022;50(1):65–75.
Sturm S, et al. A phase 1 healthy male volunteer single escalating dose study of the pharmacokinetics and pharmacodynamics of risdiplam (RG7916, RO7034067), a SMN2 splicing modifier. Br J Clin Pharmacol. 2019;85(1):181–93.
Poirier A, et al. Risdiplam distributes and increases SMN protein in both the central nervous system and peripheral organs. Pharmacol Res Perspect. 2018;6(6): e00447.
Koukouritaki SB, et al. Human hepatic flavin-containing monooxygenases 1 (FMO1) and 3 (FMO3) developmental expression. Pediatr Res. 2002;51(2):236–43.
Shimizu M, et al. Developmental variations in metabolic capacity of flavin-containing mono-oxygenase 3 in childhood. Br J Clin Pharmacol. 2011;71(4):585–91.
Xu M, et al. Genetic and nongenetic factors associated with protein abundance of flavin-containing monooxygenase 3 in human liver. J Pharmacol Exp Ther. 2017;363(2):265–74.
Cashman JR, Zhang J. Human flavin-containing monooxygenases. Annu Rev Pharmacol Toxicol. 2006;46:65–100.
Bloom AJ, et al. Effects upon in-vivo nicotine metabolism reveal functional variation in FMO3 associated with cigarette consumption. Pharmacogenet Genom. 2013;23(2):62–8.
Overby LH, Carver GC, Philpot RM. Quantitation and kinetic properties of hepatic microsomal and recombinant flavin-containing monooxygenases 3 and 5 from humans. Chem Biol Interact. 1997;106(1):29–45.
Parte P, Kupfer D. Oxidation of tamoxifen by human flavin-containing monooxygenase (FMO) 1 and FMO3 to tamoxifen-N-oxide and its novel reduction back to tamoxifen by human cytochromes P450 and hemoglobin. Drug Metab Dispos. 2005;33(10):1446–52.
Cleary Y, et al. Model-based drug–drug interaction extrapolation strategy from adults to children: risdiplam in pediatric patients with spinal muscular atrophy. Clin Pharmacol Ther. 2021;110(6):1547–57.
Upreti VV, Wahlstrom JL. Meta-analysis of hepatic cytochrome P450 ontogeny to underwrite the prediction of pediatric pharmacokinetics using physiologically based pharmacokinetic modeling. J Clin Pharmacol. 2016;56(3):266–83.
Cashman JR. Role of flavin-containing monooxygenase in drug development. Expert Opin Drug Metab Toxicol. 2008;4(12):1507–21.
US FDA. Drug Development and Drug Interactions: Table of Substrates, Inhibitors and Inducers. https://www.fda.gov/drugs/drug-interactions-labeling/drug-development-and-drug-interactions-table-substrates-inhibitors-and-inducers. Accessed Nov 2022.
Salem F, Rostami-Hodjegan A, Johnson TN. Do children have the same vulnerability to metabolic drug-drug interactions as adults? A critical analysis of the literature. J Clin Pharmacol. 2013;53(5):559–66.
Edginton AN, et al. A mechanistic approach for the scaling of clearance in children. Clin Pharmacokinet. 2006;45(7):683–704.
Johnson TN, Rostami-Hodjegan A, Tucker GT. Prediction of the clearance of eleven drugs and associated variability in neonates, infants and children. Clin Pharmacokinet. 2006;45(9):931–56.
Salem F, et al. A re-evaluation and validation of ontogeny functions for cytochrome P450 1A2 and 3A4 based on in vivo data. Clin Pharmacokinet. 2014;53(7):625–36.
Lang J, et al. Impact of hepatic CYP3A4 ontogeny functions on drug–drug interaction risk in pediatric physiologically-based pharmacokinetic/pharmacodynamic modeling: critical literature review and ivabradine case study. Clin Pharmacol Ther. 2021;109(6):1618–30.
Johnson TN, et al. Enterocytic CYP3A4 in a paediatric population: developmental changes and the effect of coeliac disease and cystic fibrosis. Br J Clin Pharmacol. 2001;51(5):451–60.
Gabrielsson J, Weiner D. Pharmacokinetic–pharmacodynamic data analysis: concepts and applications. 1997.
Richards FJ. A flexible growth function for empirical use. J Exp Bot. 1959;10(29):290–300.
Barter ZE, et al. Scaling factors for the extrapolation of in vivo metabolic drug clearance from in vitro data: reaching a consensus on values of human microsomal protein and hepatocellularity per gram of liver. Curr Drug Metab. 2007;8(1):33–45.
Leeder JS, et al. Ontogeny of scaling factors for pediatric physiology-based pharmacokinetic modeling and simulation: microsomal protein per gram of liver. Drug Metab Dispos. 2022;50(1):24–32.
Anderson BJ, Holford NH. Mechanism-based concepts of size and maturity in pharmacokinetics. Annu Rev Pharmacol Toxicol. 2008;48:303–32.
Brussee JM, et al. Characterization of intestinal and hepatic CYP3A-mediated metabolism of midazolam in children using a physiological population pharmacokinetic modelling approach. Pharm Res. 2018;35(9):182.
Nagar S et al. Enzyme kinetics in drug metabolism: fundamentals and applications. Methods Mol Biol. 2021;2342:551–93.
Shimizu M, et al. Complex mechanism underlying transcriptional control of the haplotyped flavin-containing monooxygenase 3 (FMO3) gene in Japanese: different regulation between mutations in 5′-upstream distal region and common element in proximal region. Drug Metab Pharmacokinet. 2008;23(1):54–8.
Edginton AN, Schmitt W, Willmann S. Development and evaluation of a generic physiologically based pharmacokinetic model for children. Clin Pharmacokinet. 2006;45(10):1013–34.
Johnson TN, Jamei M, Rowland-Yeo K. How does in vivo biliary elimination of drugs change with age? Evidence from in vitro and clinical data using a systems pharmacology approach. Drug Metab Dispos. 2016;44(7):1090–8.
Yeung CK, Adman ET, Rettie AE. Functional characterization of genetic variants of human FMO3 associated with trimethylaminuria. Arch Biochem Biophys. 2007;464(2):251–9.
US FDA. Center for Drug Evaluation and Research. Application Number: 213535Orig1s000 Clinical Pharmacology Review(s). 2020. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/213535Orig1s000ClinPharmR.pdf. Accessed May 2022.
Galetin A, et al. Prediction of time-dependent CYP3A4 drug-drug interactions: impact of enzyme degradation, parallel elimination pathways, and intestinal inhibition. Drug Metab Dispos. 2006;34(1):166–75.
Obach RS. Predicting drug-drug interactions from in vitro drug metabolism data: challenges and recent advances. Curr Opin Drug Discov Devel. 2009;12(1):81–9.
Acknowledgements
The authors would like to thank all the SMA patients and families who participated in the risdiplam programme, the patient groups, and the clinical trial sites and staff for their support, as well as Valérie Cosson for the scientific discussions on the initial PPK modelling of risdiplam.
Author information
Authors and Affiliations
Contributions
YC, AG and MG wrote the manuscript; YC, MG and AG designed the research; YC, HK, KH, PG, KO, HSB, NF, LA, AG and MG performed the research; and YC and MG analysed the data.
Corresponding authors
Ethics declarations
Conflict of interest
Yumi Cleary, Heidemarie Kletzl, Paul Grimsey, Katja Heinig, Hanna E. Silber Baumann, Nicolas Frey and Michael Gertz are employees of F. Hofmann-La Roche, Ltd, and hold stocks/stock options with F. Hoffmann-La Roche, Ltd. Kayode Ogungbenro, Leon Aarons and Aleksandra Galetin declared no competing interests for this work.
Ethics approval
All relevant study documents were approved by the Institutional Review Board and all subjects or their guardians signed the informed consent prior to enrolment. The studies were conducted in full conformance with the principles of the Declaration of Helsinki and its later amendments or comparable ethical standards.
Consent to participate
Informed consent was obtained from all individual participants included in the studies discussed.
Funding
This study was funded by F. Hoffmann-La Roche Ltd. Open access publication of this manuscript was sponsored by F. Hoffmann-La Roche, Ltd.
Data availability
Qualified researchers may request access to individual patient-level data through the clinical study data request platform (https://vivli.org/). Further details on Roche’s criteria for eligible studies are available at https://vivli.org/members/ourmembers/. For further details on Roche’s Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see https://www.roche.com/research_and_development/who_we_are_how_we_work/research_and_clinical_trials/our_commitment_to_data_sharing.htm.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Cleary, Y., Kletzl, H., Grimsey, P. et al. Estimation of FMO3 Ontogeny by Mechanistic Population Pharmacokinetic Modelling of Risdiplam and Its Impact on Drug–Drug Interactions in Children. Clin Pharmacokinet 62, 891–904 (2023). https://doi.org/10.1007/s40262-023-01241-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-023-01241-7