
Abstract
Background and Objective
Dapagliflozin, a sodium-glucose co-transporter inhibitor, was originally developed as an oral glucose-lowering drug for the treatment of type 2 diabetes mellitus. Emerging data suggest that cardiovascular and kidney benefits extend to patients without diabetes. Limited pharmacological data are, however, available in patients without diabetes. We aimed to characterise the pharmacokinetic profile of dapagliflozin in patients with chronic kidney disease without type 2 diabetes.
Methods
Plasma samples were collected in a randomised, placebo-controlled, double-blind, cross-over trial (DIAMOND, NCT03190694, n = 53) that assessed the effects of 10 mg of dapagliflozin in patients with a glomerular filtration rate ≥ 25 mL/min/1.73 m2 and proteinuria > 500 mg/day. Mixed-effects models were used to develop a pharmacokinetic model and to evaluate the association between plasma exposure and response.
Results
Plasma concentrations (n = 430 observations) from 48 patients (mean age 50.8 years, mean glomerular filtration rate 57.9 mL/min/1.73 m2, median proteinuria 1115 mg/24 h) were best described using a two-compartment model with first-order elimination. Apparent clearance and volume of distribution were 11.7 (95% confidence interval 10.7–12.7) L/h and 44.9 (95% confidence interval 39.0–50.9) L, respectively. Median dapagliflozin plasma exposure was 740.9 ng h/mL (2.5th–97.5th percentiles: 434.0–1615.3). Plasma exposure increased with decreasing kidney function. Every 100-ng h/mL increment in dapagliflozin plasma exposure was associated with a decrease in the urinary albumin:creatinine ratio (β = − 2.8%, p = 0.01), glomerular filtration rate (β = − 0.5 mL/min/1.73 m2, p < 0.01) and systolic blood pressure (β = − 0.4 mmHg, p = 0.03).
Conclusions
The dapagliflozin plasma concentration–time profile in patients with non-diabetic kidney disease appears similar to the profile of patients with diabetic kidney disease described in the literature. Furthermore, the plasma exposure was associated with changes in risk markers for kidney disease.
Similar content being viewed by others

1 Introduction
Sodium-glucose co-transporter 2 (SGLT2) inhibitors were originally developed as oral anti-diabetic drugs that lower plasma glucose and glycated haemoglobin by promoting urinary glucose excretion [1, 2]. In addition to improving glycaemic control, SGLT2 inhibitors exert beneficial effects on risk markers for kidney disease, such as body weight, systolic blood pressure and albuminuria [3, 4]. Large outcome trials have shown that SGLT2 inhibitors improve cardiovascular outcomes and delay the progression of kidney function decline in patients with type 2 diabetes mellitus and chronic kidney disease [5, 6]. These clinical benefits are unlikely explained by improvements in glycaemic control alone and are thought to be the result of direct effects on kidney and systemic vascular haemodynamics [7]. This suggests that SGLT2 inhibitors may also have beneficial effects in patients without diabetes with cardiovascular or kidney disease.
As SGLT2 inhibitors were originally developed for the treatment of type 2 diabetes, earlier pharmacokinetic and dose-finding studies focused on characterising the pharmacokinetics and pharmacodynamics of SGLT2 inhibitors in patients with type 2 diabetes with or without kidney disease [8]. These studies revealed that the plasma exposure of SGLT2 inhibitors increased in patients with impaired kidney function [9,10,11,12]. In contrast, pharmacodynamic effects of SGLT2 inhibitors on glucose excretion were attenuated in patients with impaired kidney function due to less glucose filtration [9,10,11,12]. The effects on body weight and blood pressure appear to be preserved [13].
Emerging data suggest that the benefits of SGLT2 inhibitors on kidney outcomes likely extend to patients without diabetes as well [14,15,16,17]. The DAPA-HF (Dapagliflozin And Prevention of Adverse Outcomes in Heart Failure) and DAPA-CKD (Dapagliflozin And Prevention of Adverse Outcomes in Chronic Kidney Disease) trials assessed the effects of the SGLT2 inhibitor dapagliflozin in broad cohorts of patients with heart failure or chronic kidney disease, respectively [15, 17]. Patients with or without type 2 diabetes participated in both trials [18]. Given the favourable results of the aforementioned outcome trials, it is likely that SGLT2 inhibitors will be prescribed in a large cohort of non-diabetic patients. In the design of these trials, it was assumed that the pharmacokinetic profile of SGLT2 inhibitors in patients without diabetes is similar to that in patients with diabetes. However, empirical data confirming this assumption are lacking.
We therefore aimed to characterise the pharmacokinetic profile of the SGLT2 inhibitor dapagliflozin in patients with non-diabetic kidney disease. Subsequently, we investigated the association between dapagliflozin plasma exposure and several risk markers for kidney disease.
2 Methods
2.1 Study Design and Patient Population
Data were used from the “Effects of the SGLT2 inhibitor dapagliflozin on proteinuria in non-diabetic patients with chronic kidney disease” (DIAMOND) trial (NCT03190694), a randomised, placebo-controlled, double-blind, cross-over trial that assessed the kidney protective effects of dapagliflozin in non-diabetic patients with albuminuria. The study design and primary results have been reported elsewhere [19]. In short, the DIAMOND trial enrolled 53 participants with non-diabetic kidney disease, characterised by 24-h urinary protein excretion > 500 mg/day and ≤ 3500 mg/day, and an estimated glomerular filtration rate (eGFR) ≥ 25 mL/min/1.73 m2. Participants had to be treated with a stable dose of an angiotensin-converting enzyme inhibitor or angiotensin II receptor blocker for at least 4 weeks prior to enrolment. Participants were randomly assigned, in a cross-over design, to placebo followed by dapagliflozin 10 mg once daily, or dapagliflozin 10 mg once daily followed by placebo. Each treatment period lasted 6 weeks, followed by a 6-week wash-out period to avoid carry-over effects. The primary endpoint of the trial was 24-h proteinuria and secondary endpoints included body weight, measured glomerular filtration rate (mGFR), systolic blood pressure and urinary albumin-to-creatinine ratio (UACR). The study was performed in accordance with the Declaration of Helsinki and good clinical practice guidelines and participants gave their written informed consent before any study-specific procedure commenced.
2.2 Measurements
Twenty-four-hour urine was collected to monitor proteinuria at the start and end of each treatment period. Body weight and systolic blood pressure were recorded at every visit to the clinic. Measured glomerular filtration rate was estimated by determining the plasma clearance of non-radioactive iohexol at the beginning and end of each treatment period. At the end of the treatment period, during GFR measurement, plasma samples of dapagliflozin were collected pre-dose, and every 30 min for 4 h after administration of dapagliflozin or placebo. Actual sampling and dosing times were recorded. The plasma concentration of dapagliflozin was measured using a liquid chromatography–tandem mass spectrometry method, which has been described elsewhere [20]. This bioanalytical method was validated for selectivity, linearity, accuracy and precision, dilution integrity, stability and recovery. The accuracy was between 94.6 and 101.0% and precision (coefficient of variation) was between 0.0 and 13.7%.
2.3 Estimation of Individual Exposure to Dapagliflozin
A population pharmacokinetic model was used to estimate individual plasma exposure to dapagliflozin. Non-linear mixed-effects models were used to develop this population pharmacokinetic model. Model development was conducted using NONMEM version 7.3.0 (ICON Development Solutions, Ellicott City, MD, USA).
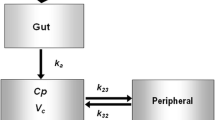
Different structural models with linear absorption and elimination processes were evaluated, including one- and two-compartment models with and without a lag time. Furthermore, the inclusion of transit compartments in the model to describe the absorption phase was also explored. A log-normal distribution was assumed for the inclusion of random effects in the stochastic model. Covariance between random effects was also evaluated. Additive, proportional and combined error models were explored to describe the residual variability. Covariate screening was performed for age, sex, race, ethnicity, eGFR, mGFR, body weight and region. We used correlation matrices of the empirical Bayes estimates of the parameters vs covariates to evaluate potential relationships. For discrete covariates, separate population parameters were estimated. For body weight, allometric scaling normalised by 70 kg was explored and, for other continuous covariates, the covariate was median normalised and a power coefficient was estimated.
First-order conditional estimation with interaction was used to obtain model parameters. Model selection and evaluation were based on the minimum objective function value (MOFV), standard goodness-of-fit plots, condition number, residual standard error of parameter estimates, and coefficient of variation of the random effects representing residual and random variability [21]. The predictive performance of the model was evaluated using a visual predictive check.
2.4 Evaluation of the Association between Exposure and Kidney Response
Risk markers of interest were proteinuria, UACR, mGFR, systolic blood pressure and body weight, which are well-known risk markers for progression of kidney disease. The individual change from baseline was estimated for all risk markers in both the placebo as well as the active treatment period. For proteinuria and UACR, the change from baseline was log-transformed to approximate a normal distribution.
Using the population pharmacokinetic model, the plasma exposure, defined as the area under the plasma concentration–time curve (AUC0–inf), was estimated by dividing the 10-mg dose by the individual apparent clearance parameter. The association between exposure to dapagliflozin, in terms of AUC0–inf, and response was investigated using linear mixed-effects models. A random intercept model was fitted to the data to estimate the placebo response of each individual patient, which was compared to a random intercept model including AUC0–inf as fixed effect. A model comparison was performed using a likelihood ratio test, which assumes a chi-square distribution. A significant increase in the maximum likelihood indicates that the addition of AUC0–inf to the model explains residual and/or between-patient variability. Furthermore, a t test was performed to evaluate whether the fixed regression coefficient of AUC0–inf was significantly different from zero. All linear mixed-effects models were fitted using full maximum-likelihood estimation. The linear mixed-effects models were fitted using the lme function of the nlme package (version nlme_3.1-131) in R version 3.4.2 (R Foundation for Statistical Computing, Vienna, Austria).
3 Results
3.1 Estimation of Plasma Exposure
In total, 477 plasma samples were collected in 51 of the 53 patients in the active treatment period. Of these, 47 samples were excluded based on: missing sampling times (n = 27 samples), missing dosing time (n = 9 samples), patient did not take medication during the study visit (n = 9 samples) and dapagliflozin concentration below the lower limit of quantification (n = 2 samples). This resulted in the inclusion of 430 samples from 48 patients in the population pharmacokinetic analysis. The demographics of patients included in the population pharmacokinetic analysis are presented in Table 1.
A two-compartment model with first-order elimination best described the plasma-concentration–time profile of dapagliflozin (Table 2). A number of transit compartments were estimated in the model to adequately describe the absorption phase. The stochastic model included interindividual variability on multiple parameters of the structural model. The residual variability was best described assuming a proportional error model. Body weight was implemented in the model assuming allometric scaling with fixed power coefficients, which significantly improved the model fit (− 26.9 ΔMOVF). Furthermore, kidney function significantly influenced the pharmacokinetics of dapagliflozin. The mGFR explained variability in the apparent clearance of dapagliflozin from the central compartment and reduced MOFV by − 28.1. In comparison, the eGFR also explained variability in the apparent clearance of dapagliflozin from the central compartment, but the improvement in model fit was less compared with mGFR (MOFV changed by − 19.3). Therefore, mGFR was included in the final model (ΔMOFV mGFR vs eGFR = 8.8, p < 0.01).
The population parameters of the pharmacokinetic model were estimated with good precision (relative standard error < 23.6%). The condition number of the model was 5.4, indicating that the model was stable. Furthermore, the population pharmacokinetic model was able to capture both the individual and central trend of the data (Fig. 1). Shrinkage was in general acceptable, but a relatively high shrinkage was observed for the number of transit compartments (42.2%).

Visual predictive check of the dapagliflozin population pharmacokinetic model. The points represent the observations of dapagliflozin plasma concentrations. Solid and dashed lines represent the observed 10th, 50th and 90th percentiles for all observations and the shaded area represents the 95% confidence interval for the 10th, 50th and 90th percentiles of the model predictions
3.2 Influence of Covariates on Plasma Exposure
The population parameters of the central compartment were 11.7 (95% confidence interval 10.7–12.7) L/h for apparent clearance and 44.9 (39.0–50.9) L for the apparent volume of distribution. The median dapagliflozin AUC0–inf after a 10-mg dose was estimated to be 740.9 ng h/mL (2.5th–97.5th percentiles: 434.0–1615.3). Dapagliflozin exposure was higher in patients with impaired kidney function and in patients with lower body weights. The predicted median dapagliflozin AUC0–inf range was from 539.2 (483.5–600.4) ng h/mL in participants of 70 kg with mGFR 90 mL/min/1.73 m2 to 1566.1 (1348.4–1855.4) ng h /mL in participants of 70 kg with mGFR 15 mL/min/1.73 m2 (Fig. 2). The predicted median dapagliflozin AUC0–inf range was from 1100.3 (1016.1–1204.1) ng h/mL in participants of 50 kg with mGFR 50 mL/min/1.73 m2 to 483.0 (445.5–527.8) ng h/mL in participants of 150 kg with mGFR 50 mL/min/1.73 m2 (Fig. 2).

Influence of kidney function and body weight on dapagliflozin plasma exposure. The points represent the estimated plasma exposure (area under the plasma–concentration time curve [AUC0–inf]) per individual normalised by the estimated plasma exposure (AUC0–inf) of a 70-kg individual (left plot) or normalised by a measured glomerular filtration rate (GFR) of 50 mL/min/1.73 m2 (right plot). The line and shaded area represent the population mean and 95% confidence interval
3.3 Evaluation of the Association between Exposure and Kidney Response
The individual change from baseline for all risk markers in both the placebo as the active treatment period is displayed in Table 3. Figure 3 shows the exposure–response relationship between dapagliflozin and UACR, mGFR, systolic blood pressure and body weight. It was estimated that every 100-ng h/mL increment in dapagliflozin AUC0–inf is associated with a decrease in UACR (β = − 2.79 %, p = 0.01), mGFR (β = − 0.54 mL/min per 1.73 m2, p < 0.01), systolic blood pressure (β = − 0.39 mmHg, p = 0.03), body weight (β = − 0.14 kg, p = 0.07) and proteinuria (β = 0.00%, p = 0.69).

Relationship between exposure and response at week 6. Observations (black circle), reflected as change from baseline, are plotted vs the area under the plasma concentration–time curve (AUC0–inf). The dotted gray lines represent the individual difference between placebo and active treatment vs AUC0–inf. The dotted blue line represents the population difference between placebo and active treatment vs AUC0–inf. GFR glomerular filtration rate
4 Discussion
We developed a population pharmacokinetic model that adequately described the individual plasma concentration–time profile of dapagliflozin in patients with non-diabetic kidney disease. We found that plasma exposure to dapagliflozin was higher in patients with impaired kidney function and in patients with relatively low body weight. Furthermore, we demonstrated that individual dapagliflozin plasma exposure was associated with changes in UACR, mGFR and systolic blood pressure.
As there is increasing interest in the use of dapagliflozin and other SGLT2 inhibitors in patients without diabetes with cardiovascular or kidney disease, it is important to characterise the plasma concentration–time profile of dapagliflozin in patients without diabetes and assess whether this profile is comparable to patients with diabetes. A population pharmacokinetic model for dapagliflozin in patients with type 2 diabetes and chronic kidney disease has been described by van der Walt et al., who reported that a two-compartment structural model, with first-order elimination and usage of multiple transit compartments for the absorption phase, best described the data [22]. Similar to the model of van der Walt et al., the individual plasma concentration–time profiles in patients without diabetes were also best described using a two-compartment model with first-order elimination, and multiple transit absorption compartments. Additionally, the estimated model parameters of the structural model are in a similar range (apparent clearance 11.7 L/h vs approximately 10.5 L/h in the model of van der Walt et al.), indicating that both models are quite comparable. Indeed, the mean estimated exposure to dapagliflozin that we found with our model in patients without diabetes is similar to the estimated exposure reported by van der Walt et al. in diabetic patients [22]. It can therefore be concluded that the plasma concentration–time profile of dapagliflozin in patients without diabetes is comparable to those with diabetes.
The variability between patients in the individual plasma concentration–time profiles after administration of 10 mg of dapagliflozin was high. The between-patient variability in the plasma concentration–time profile was in part explained by differences in kidney function and body weight between patients. The model predicted that dapagliflozin plasma exposure nearly doubled in patients with mGFR of 30 mL/min/1.73 m2 and nearly tripled in patients with mGFR of 15 mL/min/1.73 m2 compared with patients with normal kidney function. These estimates are consistent with pharmacokinetic studies in patients with type 2 diabetes and kidney impairment [11]. Kidney function and body weight were also identified in the population pharmacokinetic model of van der Walt et al. as significant predictors for individual plasma exposure [22]. An advantage of our study was that we measured GFR by iohexol plasma clearance whereas other studies have used less precise serum creatinine-based equations to estimate GFR. Measured glomerular filtration rate is considered to be a more accurate marker of the actual kidney function than eGFR and thus provides a better estimate of the influence of kidney function on plasma exposure [23]. Indeed, in our model, dapagliflozin plasma concentrations were better described with mGFR compared with eGFR.
In the present study, we demonstrate that the plasma exposure to dapagliflozin is associated with beneficial changes in risk markers for kidney disease in patients with non-diabetic kidney disease. A higher plasma exposure to dapagliflozin is associated with a more pronounced decrease in UACR, mGFR and systolic blood pressure. Additionally, a trend for a decrease in body weight with increased exposure was observed. These findings are in keeping with those previously reported in patients with type 2 diabetes and suggests dapagliflozin has an effect on these risk markers [24, 25]. Dapagliflozin did not decrease proteinuria in the DIAMOND trial [19]. Between-patient variability in proteinuria change was also not associated with dapagliflozin plasma exposure. In contrast, the statistically significant association between exposure and UACR response suggests that the reduction in UACR in the DIAMOND trial is real. The lack of an association between plasma exposure and proteinuria response, in contrast with the association between plasma exposure and albuminuria response, suggests that albuminuria might be a more suitable risk marker for detecting drug effects in patients with kidney disease.
Based on our models, patients with impaired kidney function have a higher plasma exposure to dapagliflozin and are therefore expected to have more pronounced effects on risk markers for kidney disease as compared with patients without impaired kidney function. Dapagliflozin is filtered by the kidney and binds to the SGLT2 transporter located in the apical membrane of the proximal tubule [26]. The total amount of filtered dapagliflozin decreases in patients with impaired kidney function, which could result in a decrease in dapagliflozin concentration in the kidney. However, patients with impaired kidney function have fewer numbers of functioning nephrons, which balances the decrease in filtered dapagliflozin and may even lead to an increase in dapagliflozin in the proximal tubule. Furthermore, dapagliflozin is metabolised in both the liver and kidney to pharmacologically inactive dapagliflozin 3-O-glucuronide by UGT1A9. The ratio between dapagliflozin and its metabolite is reduced in patients with kidney impairment [22, 27]. This suggests that dapagliflozin metabolism in the kidney is also decreased in patients with kidney disease, which seems plausible as UGT1A9 expression is eight-fold higher in the kidney as compared with the liver [22]. The concentration of dapagliflozin could therefore locally be increased in patients with kidney impairment, which could explain the more pronounced effects of dapagliflozin on risk markers for kidney disease. Unfortunately, we were not able to measure dapagliflozin 3-O-glucuronide concentrations, thus this possibility needs to be further explored in future studies.
This study has a number of limitations. First, all participants were treated with dapagliflozin 10 mg. Therefore, our analysis is limited to explaining variability in kidney response between patients and cannot be used to make any dosing recommendations for patients with impaired kidney function. Second, model parameters were estimated with reasonable precision, but shrinkages in the parameters for the transit compartment were relatively high. This was also observed for patients with diabetic kidney disease [22]. This is most likely caused by a relatively low amount of plasma samples that were collected in the absorption phase of patients that demonstrated a relatively fast absorption. Consequently, the model should not be used to predict the maximum concentration. Third, no data were available of patients with very low kidney function (mGFR ≤ 20 mL/min/1.73 m2). The predicted effects in patients with severe kidney impairment might therefore be overestimated in our model. Additional data are needed to draw definitive conclusions for this subgroup of patients. Fourth, we did not measure plasma metabolites nor urine concentrations of both the parent and metabolite and were thus unable to estimate absolute bioavailability. Finally, it should be noted that our pharmacokinetic model has not been externally validated.
5 Conclusions
We developed a pharmacokinetic model that adequately estimated the plasma exposure of the SGLT2 inhibitor dapagliflozin in patients with non-diabetic kidney disease. The plasma concentration–time profile of dapagliflozin in patients with non-diabetic kidney disease appears to be comparable to patients with diabetic kidney disease. Furthermore, dapagliflozin plasma exposure was associated with several risk markers for kidney disease. These findings suggest that the clinically used doses of 10 mg of dapagliflozin can also be used in patients without diabetes.
References
List JF, Woo V, Morales E, Tang W, Fiedorek FT. Sodium-glucose cotransport inhibition with dapagliflozin in type 2 diabetes. Diabetes Care. 2009;32:650–7.
Zhang L, Feng Y, List J, Kasichayanula S, Pfister M. Dapagliflozin treatment in patients with different stages of type 2 diabetes mellitus: effects on glycaemic control and body weight. Diabetes Obes Metab. 2010;12:510–6.
Heerspink HJ, Perkins BA, Fitchett DH, Husain M, Cherney DZ. Sodium glucose cotransporter 2 inhibitors in the treatment of diabetes mellitus: cardiovascular and kidney effects, potential mechanisms, and clinical applications. Circulation. 2016;134:752–72.
Inzucchi SE, Zinman B, Fitchett D, et al. How does empagliflozin reduce cardiovascular mortality? Insights from a mediation analysis of the EMPA-REG OUTCOME trial. Diabetes Care. 2018;41:356–63.
Zelniker TA, Wiviott SD, Raz I, et al. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: a systematic review and meta-analysis of cardiovascular outcome trials. Lancet. 2019;393:31–9.
Perkovic V, Jardine MJ, Neal B, et al. Canagliflozin and renal outcomes in type 2 diabetes and nephropathy. N Engl J Med. 2019;380:2295–306.
Heerspink HJ, Desai M, Jardine M, Balis D, Meininger G, Perkovic V. Canagliflozin slows progression of renal function decline independently of glycemic effects. J Am Soc Nephrol. 2017;28:368–75.
Kasichayanula S, Liu X, Lacreta F, Griffen SC, Boulton DW. Clinical pharmacokinetics and pharmacodynamics of dapagliflozin, a selective inhibitor of sodium-glucose co-transporter type 2. Clin Pharmacokinet. 2014;53:17–27.
Devineni D, Curtin CR, Marbury TC, et al. Effect of hepatic or renal impairment on the pharmacokinetics of canagliflozin, a sodium glucose co-transporter 2 inhibitor. Clin Ther. 2015;37(610–28):e4.
Ferrannini E, Veltkamp SA, Smulders RA, Kadokura T. Renal glucose handling: impact of chronic kidney disease and sodium-glucose cotransporter 2 inhibition in patients with type 2 diabetes. Diabetes Care. 2013;36:1260–5.
Kasichayanula S, Liu X, Pe Benito M, et al. The influence of kidney function on dapagliflozin exposure, metabolism and pharmacodynamics in healthy subjects and in patients with type 2 diabetes mellitus. Br J Clin Pharmacol. 2013;76:432–44.
Macha S, Mattheus M, Halabi A, Pinnetti S, Woerle HJ, Broedl UC. Pharmacokinetics, pharmacodynamics and safety of empagliflozin, a sodium glucose cotransporter 2 (SGLT2) inhibitor, in subjects with renal impairment. Diabetes Obes Metab. 2014;16:215–22.
Scheen AJ. Pharmacokinetic/pharmacodynamic properties and clinical use of SGLT2 inhibitors in non-Asian and Asian patients with type 2 diabetes and chronic kidney disease. Clin Pharmacokinet. 2020;59:981–94.
Petrie MC, Verma S, Docherty KF, et al. Effect of dapagliflozin on worsening heart failure and cardiovascular death in patients with heart failure with and without diabetes. JAMA. 2020;323:1353–68.
Heerspink HJL, Stefánsson BV, Correa-Rotter R, et al. Dapagliflozin in patients with chronic kidney disease. N Engl J Med. 2020;383:1436–46.
Packer M, Anker SD, Butler J, et al. Cardiovascular and renal outcomes with empagliflozin in heart failure. N Engl J Med. 2020;83:1413–24.
McMurray JJV, Solomon SD, Inzucchi SE, et al. Dapagliflozin in patients with heart failure and reduced ejection fraction. N Engl J Med. 2019;381:1995–2008.
Heerspink HJL, Stefansson BV, Chertow GM, et al. Rationale and protocol of the Dapagliflozin And Prevention of Adverse outcomes in Chronic Kidney Disease (DAPA-CKD) randomized controlled trial. Nephrol Dial Transplant. 2020;35:274–82.
Cherney DZI, Dekkers CCJ, Barbour SJ, et al. Effects of the SGLT2 inhibitor dapagliflozin on proteinuria in non-diabetic patients with chronic kidney disease (DIAMOND): a randomised, double-blind, crossover trial. Lancet Diabetes Endocrinol. 2020;8:582–93.
van der Aart-van der Beek AB, Wessels AMA, Heerspink HJL, Touw DJ. Simple, fast and robust LC–MS/MS method for the simultaneous quantification of canagliflozin, dapagliflozin and empagliflozin in human plasma and urine. J Chromatogr B Anal Technol Biomed Life Sci. 2020;1152:122257.
Mould DR, Upton RN. Basic concepts in population modeling, simulation, and model-based drug development-part 2: introduction to pharmacokinetic modeling methods. CPT Pharmacometr Syst Pharmacol. 2013;2:e38.
van der Walt JS, Hong Y, Zhang L, Pfister M, Boulton DW, Karlsson MO. A nonlinear mixed effects pharmacokinetic model for dapagliflozin and dapagliflozin 3-O-glucuronide in renal or hepatic impairment. CPT Pharmacometr Syst Pharmacol. 2013;2:e42.
Porrini E, Ruggenenti P, Luis-Lima S, et al. Estimated GFR: time for a critical appraisal. Nat Rev Nephrol. 2019;15:177–90.
Koomen JV, Stevens J, Heerspink HJL. Exposure–response relationships of dapagliflozin on cardiorenal risk markers and adverse events: a pooled analysis of 13 phase II/III trials. Br J Clin Pharmacol. 2020. https://doi.org/10.1111/bcp.14318.
Kroonen MYAM, Koomen JV, Petrykiv SI, Laverman GD, Heerspink HJL, Stevens J. Exposure–response relationships for the sodium-glucose co-transporter-2 inhibitor dapagliflozin with regard to renal risk markers. Diabetes Obes Metab. 2020;22:916–21.
Ghezzi C, Yu AS, Hirayama BA, et al. Dapagliflozin binds specifically to sodium-glucose cotransporter 2 in the proximal renal tubule. J Am Soc Nephrol. 2017;28:802–10.
Committee for Medicinal Products for Human Use (CHMP). European public assessment report Forxiga dapagliflozin (Procedure No.: EMEA/H/C/002322, EMA/689976/2012). European medicines Agency (EmA), Amsterdam.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The DIAMOND trial was funded by Astra Zeneca.
Conflict of interest
Annemarie B. van der Aart-van der Beek, Abdul Halim Abdul Gafor, Claire C.J. Dekkers, Daniel C. Cattran, Jasper Stevens, Jeroen V. Koomen, Qiang Li, Sean J. Barbour and Sunita Singh have no conflicts of interest that are directly relevant to the content of this article. Hiddo J.L. Heerspink is a consultant to Abbvie, AstraZeneca, Boehringer Ingelheim, Bayer, Chinook, CSL Behring, Gilead, Janssen, Merck, Mundipharma, Mitsubishi Tanabe, Novo Nordisk and Retrophin. He received research support from AstraZeneca, Abbvie, Boehringer Ingelheim and Janssen. David W. Boulton is an employee and shareholder of AstraZeneca. David Z.I. Cherney has received honoraria from Boehringer Ingelheim-Lilly, Merck, AstraZeneca, Sanofi, Mitsubishi-Tanabe, Abbvie, Janssen, Bayer, Prometic, BMS and Novo-Nordisk and has received operational funding for clinical trials from Boehringer Ingelheim-Lilly, Merck, Janssen, Sanofi, AstraZeneca and Novo-Nordisk. Ron T. Gansevoort has consulting agreements with AstraZeneca, Bayer, Sanofi-Genzyme and Mundi Pharma; all fees are paid to his institution. Peter J. Greasley is employed by and owns shares in AstraZeneca. Gozewijn D. Laverman has received research grants and consulting fees from Sanofi and AstraZeneca, and research grants from Novo Nordisk. Marc G. Vervloet has received consulting fees from Amgen, Vifor, Fresenius Medical Care Renal Pharma, Medice, Kyowa Kirin and Astra Zeneca. Heather N. Reich has received consulting fees from Omeros and was involved in clinical trials supported by Omeros and Calliditas. Soo Kun Lim has received consulting fees or speaking honoraria from AstraZeneca, Boehringer Ingelheim, Novo Nordisk, Fresenius Kabi, Baxter and Sanofi.
Authors’ contributions
JVK analysed the data. ABvdAB, JVK and HJLH wrote the manuscript. Other authors revised the draft manuscript for important intellectual content. All authors contributed to the data collection and all authors approved the manuscript for submission.
Ethics Approval
The DIAMOND studie was performed in line with the principles of the Declaration of Helsinki. Approval was granted by the Ethics Committees of each participating center.
Consent to Participate
All DIAMOND participants provided written informed consent.
Consent for Publication
All authors approved and provied consent for publication.
Availability of Data and Material
The datasets generated during and/or analysed during the curret study are available from the corresponding author on reasonable request.
Code Availability
Not applicable.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
van der Aart-van der Beek, A.B., Koomen, J.V., Dekkers, C.C.J. et al. Evaluation of the Pharmacokinetics and Exposure–Response Relationship of Dapagliflozin in Patients without Diabetes and with Chronic Kidney Disease. Clin Pharmacokinet 60, 517–525 (2021). https://doi.org/10.1007/s40262-020-00956-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-020-00956-1