
Abstract
Background and Objective
An orodispersible tablet (ODT) formulation of morphine sulfate has been developed to provide a novel alternative for patients with severe pain requiring opioids. This formulation has been developed in a range of doses (1–30 mg), enabling relief from severe pain to be achieved and maintained with the lowest possible morphine dose for each patient. The ODT formulation is particularly suitable for patients with swallowing difficulties.
Objective
The aim of this study was to compare the pharmacokinetics and bioequivalence of the ODTs with reference formulations of morphine sulfate.
Methods
Three randomized, single-dose, laboratory-blinded, phase I, crossover studies were conducted in adult healthy volunteers under fasting conditions. The pharmacokinetics of a 30 mg morphine sulfate ODT were compared with those of equivalent doses of currently marketed oral immediate-release formulations: tablets (Sevredol®), capsules (Actiskenan®), and a solution (Oramorph®). The bioequivalence of 30 mg and 10 mg doses of the ODTs and tablets was then assessed in two further studies. Subjects were asked to complete a product appreciation questionnaire for two morphine formulations (ODT and solution).
Results
A total of 104 subjects were included across the three studies. The pharmacokinetics of the ODTs were assessed in 100 subjects and were found to be similar to those of the reference formulations. The time to maximum plasma concentration (Tmax) for the ODTs was 0.8 h, within the range observed for the reference formulations (0.75–1.25 h). Maximum plasma concentrations (Cmax) for the ODTs were 7.7 ± 2.7 ng/mL for the 10 mg dose and 26.1 ± 10.0 ng/mL for the 30 mg dose. These values were similar to those obtained for the 10 mg and 30 mg tablets (8.0 ± 2.9 ng/mL and 28.5 ± 11.9 ng/mL, respectively), and for the 30 mg capsule (29.9 ± 13.0 ng/mL). A higher Cmax was observed for the solution (37.9 ± 16.5 ng/mL). Plasma exposure to morphine (area under the plasma drug concentration-time curve [AUC]) after ODT administration was similar to that observed for the reference formulations: 39.8 ± 14.8 ng·h/mL and 115.5 ± 34.6 ng·h/mL for the 10 mg and 30 mg ODTs, versus 40.7 ± 13.5 ng·h/mL and 117.4 ± 31.5 ng·h/mL for the 10 mg and 30 mg tablets, and 121.8 ± 32.0 ng·h/mL and 121.0 ± 35.7 ng·h/mL for the 30 mg solution and capsule, respectively. Bioequivalence of the 30 mg and 10 mg ODTs and tablets, assessed in 83 patients across two studies, was demonstrated for both the Cmax and AUC from time zero to time t (AUC0–t). No serious or unexpected drug-related events were reported. A product appreciation questionnaire concluded that both ODTs and oral solution products were considered pleasant by most of the subjects.
Conclusion
The ODTs were safe, well tolerated, and showed similar pharmacokinetics to those of the reference formulations. The development of a range of doses of morphine sulfate ODTs may provide a new alternative for the oral administration of immediate-release morphine for pain management in pediatric, geriatric and adult populations with swallowing problems.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
An orodispersible tablet (ODT) formulation of morphine sulfate has recently been developed in a range of doses to provide an easy and flexible mode of administration of this opioid for severe pain management in pediatric, elderly and adult patients. |
This orodispersible formulation is particularly suitable for patients with swallowing problems. |
The current studies showed that the pharmacokinetic, safety and palatability profiles of the ODTs were similar to those of marketed products, and demonstrated the bioequivalence of two different doses of the ODT with marketed solid tablet formulations. |
1 Introduction
Morphine is well established as a first-line strong opioid in the treatment of moderate-to-severe pain [1] and its pharmacokinetic profile has been extensively studied [2]. Morphine sulfate is well absorbed and undergoes first-pass metabolism in the liver and gut. It is distributed throughout the body. Its main metabolites are morphine-3-glucoronide and morphine-6-glucoronide. Its mean plasma elimination half-life after intravenous administration is about 2 h [2]. Morphine depicts linear pharmacokinetics over a large range of doses including the 1–30 mg range [3,4,5]. A wide range of morphine formulations are included in the World Health Organization (WHO) model list of essential medicines for pain and palliative care, and for preoperative medication and sedation for short-term procedures in both children [6] and adults [7]. Morphine can be administered as oral immediate-release or modified-release tablets or capsules, as an oral solution/suspension, or as parenteral forms.
Determining the appropriate morphine dosage requires careful assessment of the type and site of origin of the pain, as well as careful consideration of the optimal treatment form. Indeed, the WHO [1] guidelines for the pharmacological and radiotherapeutic management of cancer pain in adults and adolescents specify that the goal of pain management is to reduce pain to a level that allows for a quality of life that is acceptable to the patient. The benefit of pain relief must also be balanced against the risk of adverse effects and overdose, which may result in respiratory depression.
The WHO [1], the European Society for Medical Oncology (ESMO) [8], the Centers for Disease Control and Prevention (CDC) [9] and the American Geriatrics Society [10] have made the following general recommendations for the prescription of opioids in pediatric, adult, and elderly populations: use of the least invasive route, i.e. the oral route, to reduce patient discomfort; use of the lowest effective dose; and use of a titration method to allow the lowest opioid dosage for pain control to be reached as rapidly as possible.
However, the use of solid oral drugs is not recommended in infants and young children [11], as well as in some elderly or adult patients, notably those with dysphagia, because of the risk of choking. Therefore, Ethypharm has developed an orodispersible tablet (ODT) formulation of morphine sulfate in a range of doses, including 1, 2.5, 5, 10, 20 and 30 mg formulations. The ODTs have been formulated to disperse rapidly in the mouth, or in a small quantity of water in a spoon, and then be easily swallowed. The wide range of doses was developed to allow titration to be started with the lowest possible dose, thus allowing the lowest maintenance dose for pain control to be achieved in all populations, from pediatric to geriatric patients. Notably, the 1 mg and 2.5 mg ODTs were developed to meet the needs of patients requiring low doses of morphine sulfate, as no immediate-release morphine formulations at these doses are currently available on the market. The ODTs also contain sweeteners and orange peel extract to counterbalance the bitterness of the active substance and make the taste of the ODTs acceptable for both children and adults.
Herein, we describe the results of three phase I studies that were conducted in adult healthy volunteers to compare the performance of the ODTs with that of marketed products. First, a pilot study was conducted to evaluate the pharmacokinetic properties and relative bioavailability of the 30 mg morphine sulfate ODT compared with three oral immediate-release formulations: a tablet form (Sevredol®), a capsule form (Actiskenan®) and a solution (Oramorph®). Two pivotal studies were then performed to assess the bioequivalence of the 30 mg and 5 mg morphine sulfate ODTs and the tablet form (Sevredol®). The comparative safety, tolerance and palatability of the ODTs and reference products were also assessed.
2 Methods
All studies were conducted in accordance with the ethical principles of the Declaration of Helsinki, the International Council for Harmonization (ICH) Guideline E6 for Good Clinical Practice (GCP), European regulation EU 536/2014, and the Tri-Council Policy Statement (Canada). All subjects volunteered to participate in the studies and signed an informed consent form (ICF) before screening. The study protocols and ICFs were approved by Health Canada and by an independent Institutional Review Board (Advarra). The bioequivalence studies were designed using a bracketing approach, as recommended by the guideline on the investigation of bioequivalence [12]. All samples were analyzed using bioanalytical methods validated according to the guideline on bioanalytical method validation [11] and in compliance with Good Laboratory Practice guidelines (Directives 2004/9/EC and 2004/10/EC).
2.1 Study Designs and Setting
The studies were conducted at the site of the clinical research organization Altasciences in Laval (Quebec, Canada). The pilot study was conducted from November to December 2018 using a randomized (1:1:1:1 ratio), single-dose, laboratory-blinded, four-period, four-sequence, crossover design. The two pivotal studies were conducted from July to August 2019 and during August 2019, and both used a randomized (1:1 ratio), single-dose, laboratory-blinded, two-treatment, four-period, two-sequence, fully replicated, crossover design. In all studies, assignment of the drug administration sequence was carried out using a computer-generated randomization list. All study drugs were administered under a fasting state, generating the most sensitive conditions for detecting potential differences between formulations.
2.2 Subjects
For the pilot study, it was estimated that 20 subjects were required to obtain a good estimate of intrasubject variability. Based on this estimate, the minimum number of subjects needed to meet the widened bioequivalence range with a statistical a priori power of at least 80% was about 38 for a replicate design study. To account for potential dropouts, 42 subjects were included in each of the pivotal studies. The main eligibility criteria were identical for all three studies. Eligible participants were healthy male or female subjects aged between 18 and 50 years, with a minimal body weight of 60 kg and a body mass index (BMI) within 18.5–30 kg/m2 for men and 20–30 kg/m2 for women, and who were non- or ex-smokers (for ≥180 days) with no history of opioid addiction. Women of childbearing potential had to be using an acceptable method of contraception.
2.3 Study Drugs
The following oral morphine sulfate formulations were evaluated: 30 mg and 5 mg ODTs (Ethypharm, France), Actiskenan® 30 mg immediate-release capsules (Ethypharm, France), Sevredol® 10 mg scored film-coated tablets (Bard Pharmaceutical Limited, UK), and Oramorph® 30 mg/5 mL unit dose solution (L.Molteni & C. Dei Fratelli Alitti Societa di Esercizio S.P.A., Italy). A concomitant oral opiate antagonist, naltrexone hydrochloride in 50 mg tablets (Revia®, Duramed Pharmaceuticals, Inc., USA), was also administered.
2.4 Study Procedures
Subjects were admitted to the clinical site on Day − 1. They were asked to fast overnight, for a minimum of 10 h prior to drug administration and for up to 4 h thereafter. Water was allowed, except for during the 1 h before and up to 1 h after administration, unless required for drug delivery. For the 30 mg dose studies, subjects first received a single 50 mg oral dose of naltrexone, 1 h prior to dosing, to block the subjective effects of morphine. For the 10 mg dose study, no opioid antagonist was administered. In each study, there was a 7-day washout period between treatments.
2.5 Pilot Study
On Day 0, subjects received one of the assigned products. Prior to dosing with 1 × 30 mg ODT, subjects were asked to pre-wet their mouth with 20 mL of water. The ODT was then placed onto the tongue and subjects were asked to suck the ODT until disintegration and then to swallow their saliva. The 1 × 30 mg capsule and 3 × 10 mg tablets were administered with 240 mL of water. Correct dosing of the 30 mg/5 mL solution was ensured by asking subjects to swallow the content of the single-dose vial followed by 20 mL of water used to rinse the vial. For pharmacokinetic analysis, blood samples (6 mL) were collected before naltrexone administration and at 0.08, 0.25, 0.50, 0.75, 1.00, 1.25, 1.50, 1.75, 2.00, 2.50, 3.00, 4.00, 6.00, 8.00, 12.00, 16.00 and 24.00 h following morphine administration.
2.5.1 Pivotal Studies
The ODTs (1 × 30 mg or 2 × 5 mg) and tablets (3 × 10 mg or 1 × 10 mg) were administered as described for the pilot study. It should be noted that Sevredol® is not available in a 5 mg tablet. For pharmacokinetic analysis, blood samples (6 mL) were collected before administration and at 0.08, 0.25, 0.50, 0.75, 1.00, 1.25, 1.50, 1.75, 2.00, 2.50, 3.00, 4.00, 6.00, 8.00, 12.00, 16.00, 24.00, 30.00 and 36.00 h following morphine administration.
2.6 Outcomes
The primary outcome of the pilot study was comparison of the morphine sulfate pharmacokinetic parameters of the test formulation (1 × 30 mg ODT) and the three reference formulations (3 × 10 mg tablets, 1 × 30 mg capsule and a 30 mg/5 mL solution), including analyses of the maximal plasma concentration (Cmax), plasma exposure (area under the curve from time zero to time t [AUC0–t] and from time zero to infinity [AUC0–∞]), time to maximum plasma concentration (Tmax) and half-life (T½), and estimation of the intrasubject coefficient of variation (CV). The primary outcome of the pivotal studies was the comparison of the relative bioavailability of the test formulations (1 × 30 mg or 2 × 5 mg ODT) and the reference formulations (3 × 10 mg or 1 × 10 mg tablets). Secondary outcomes included an evaluation of the palatability of the 30 mg ODT compared with that of the 30 mg/5 mL solution in the pilot study, and an evaluation of the comparative safety and tolerability of the test and reference formulations in both the pilot study and the two pivotal studies.
2.7 Bioanalytical Methods
Morphine sulfate concentrations were quantified in plasma, following solid-phase extraction, using a validated high-performance liquid chromatography (HPLC) method with tandem mass spectrometry (MS/MS) detection (AB Sciex API 5000 quadrupole mass spectrometer) in positive ion mode. A reversed-phase C18 column (50 × 2.10 mm, with 3.5 µm porosity) was used with two mobile-phase gradients (A: 20 mM NH4HCO3, pH 10.0; and B: ACN:MeOH 75:25 v/v) and a constant flow rate of 0.5 mL/min. Retention times for morphine (purity 99.6%) and morphine-D6 (purity 98.9%), used as internal standards (Cerilliant Corp., Round Rock, TX, USA), were 0.92 min and 0.88 min, respectively. Peak area ratios were used for quantification, and linearity of the calibration curve was determined using a 1/×2 weighted linear least squares (LS) regression analysis. The lower limit of quantification (LLOQ) of 50.0 pg/mL had a between-run accuracy of 91.4% and precision of 6.7%. All calibration and quality control samples were within the acceptance criteria: CV% < 15% and nominal range values within 85–115%.
2.8 Pharmacokinetic Analysis
Pharmacokinetic parameters were calculated using Phoenix WinNonlin® software, version 8.0. The main absorption and disposition parameters were calculated for individual concentration-time profiles using a noncompartmental approach with a log-linear terminal phase assumption and using the trapezoidal rule to estimate the AUC [AUC = ½ (C1 + C2) (t2 − t1)], where C is concentration and t is time. Concentrations below the LLOQ were treated as zero for all pharmacokinetic analyses. The actual time of pharmacokinetic sampling was used for the determination of all pharmacokinetic parameters. Concentration values were considered as missing if they could not be measured due to analytical or clinical issues. AUC values were not determined if fewer than three consecutive concentration values were obtained. Subjects were excluded from the pharmacokinetic analysis if their predose morphine sulfate concentration was > 5% of the Cmax value, or if no value or very low concentration values were detected.
2.9 Safety and Tolerability
The safety and tolerability of the test and reference formulations was assessed by evaluating the type, frequency, and severity of adverse events (AEs) reported during each of the three studies from date of signature of the ICF until 1 day after the last blood sample was taken for the study. AEs were defined as any untoward medical occurrence in a study subject that did not necessarily have a causal relationship with the treatment. Serious AEs (SAEs) were defined as any untoward medical occurrence that, at any treatment dose, resulted in death, risk of death, hospital care, persistent or significant disability or incapacity, or any important medical event including a congenital anomaly or birth defect. The severity of AEs/SAEs (mild, moderate or severe) was evaluated according to their impact on the usual activities of the subject. The potential relationship between the AEs/SAEs (reasonable possibility or no reasonable possibility) and any of the treatments was evaluated on the basis of a temporal relationship or the occurrence of a pharmacologically predicted event.
2.10 Palatability Assessments
Subjects participating in the pilot study evaluated the aroma, taste and texture of the 30 mg ODT and 30 mg/5 mL solution shortly after administration by completing a questionnaire containing five questions: ‘How do you rate the taste/aroma?’ (very good, good, neutral, bad, very bad); ‘How do you rate the level of bitterness?’ (not at all bitter, slightly bitter, bitter, too bitter); ‘How do you rate this type of formulation for oral administration’ (pleasant, unpleasant); ‘What do you think of the texture of the product in the mouth?’ (pleasant, unpleasant); and ‘In your opinion, in case of intense pain, would the taste or bitterness of the product prevent you from taking this treatment?’ (yes, no).
2.11 Statistical Methods
Statistical analyses were carried out using SAS® version 9.4 (generalized linear model [GLM] procedure). Pharmacokinetic parameters, safety data, and demographic variables were analyzed using descriptive statistics. Only subjects who completed all study periods were included in the pharmacokinetic analyses (pharmacokinetic populations), whereas all subjects were included in the safety analyses (safety populations). For paired comparisons, all subjects who provided evaluable data for both treatments of interest were included in the statistical analyses. An analysis of variance (ANOVA) was performed on the natural log-transformed values for Cmax, AUC0–t and AUC0–∞, as well as on the rank-transformed Tmax values. The subject (nested within the treatment sequence), treatment, sequence, and period effects were evaluated at the 5% significance level. Bioequivalence was confirmed if the 90% confidence interval (CI) for the test-to-reference geometric LS mean ratio was within 80.00–125.00% for both AUC0–t and Cmax.
3 Results
3.1 Healthy Subject Characteristics
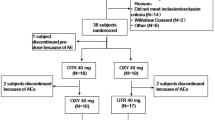
Of the 20 subjects included in the pilot study, 17 (85%) completed the study and 3 (15%) were withdrawn (consent withdrawal for personal reasons or nonappearance). Forty-two subjects were included in each of the pivotal studies. For the 30 mg dose study, 39 subjects (93%) completed the study and three subjects (7%) were withdrawn (consent withdrawal for personal reasons or nonappearance). For the 10 mg dose study, 40 subjects (95%) completed the study and two subjects (5%) were withdrawn (protocol withdrawal criterion met). The baseline demographic characteristics of the pharmacokinetic populations are presented in Table 1. Overall, 51% of the healthy subjects were male, the average BMI was 25.0 kg/m2 and the average age was 37 years (18–60).
3.2 Pharmacokinetic Analysis
3.2.1 Pilot Study
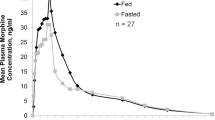
The mean plasma concentration versus time profile of the 30 mg morphine sulfate ODT was similar to those of the 3 × 10 mg tablets, 1 × 30 mg capsule and 30 mg/5 mL liquid morphine formulations, with variability observed around the Cmax (Fig. 1). Morphine exposure (AUC0–t) after administration of the ODT was 96.5 ng·h/mL. Similar AUC0–t values were obtained for the tablet, capsule and liquid morphine formulations, with mean differences in exposure below 10% (Table 2). The mean Cmax for the ODT was 28.5 ng/mL (CV = 44.0%), which was similar to the values obtained for the capsule and the tablets (mean differences < 5%) (Table 2). The Cmax for the morphine solution was about 30% higher than that for the ODT. The median Tmax was 1 h (0.5–8.0) for the ODT, 0.75 h (0.5–6.0) for the solution, 0.75 h (0.5–3.0) for the tablets, and 1.25 h (0.5–2.5) for the capsule (Table 2). Of note, two extreme values were observed for the ODT and the solution, with values of 8 h and 6 h, respectively, resulting in an upward shift of the median Tmax for these formulations (Fig. 2). Both these extreme values emanated from the same subject who suffered from nausea and emesis; however, no significant difference between ranked Tmax values was found. Morphine T½ after administration of the 30 mg ODT was 9.6 h, which was within the range of T½ values observed for the three reference formulations (8.7–10.7 h) (Table 2).

Linear profile of the mean plasma concentration versus time for morphine after administration of 30 mg of immediate-release morphine sulfate as an orodispersible tablet (ODT; 1 × 30 mg, test product, blue squares), solution (30 mg/5 mL, Oramorph®, orange circles), capsule (1 × 30 mg, Actiskenan®, grey diamonds) or tablets (3 × 10 mg, Sevredol®, green triangles) to healthy subjects (n = 17) under fasting conditions in the pilot study. Insert shows the mean plasma concentration versus time profiles to the last collection time (24 h after administration). For graph readability, one-sided error bars were represented (−SD for ODT and capsule data, and +SD for solution and tablet data). ODT orodispersible tablet, SD standard deviation

Box plots of the distribution of Tmax values for plasma morphine sulfate obtained from the pilot study. The box plots were generated using individual Tmax values obtained after the administration of (from left to right) 30 mg of immediate-release morphine sulfate as an oral dispersible tablet (ODT; 1 × 30 mg), solution (30 mg/5 mL, Oramorph®), capsule (1 × 30 mg, Actiskenan®) or tablet (3 × 10 mg, Sevredol®) to healthy subjects (n = 17) under fasting conditions in the pilot study. Boxes represent the first and third quartile together with the median line. Mean values are depicted by crosses. Minimum and maximum values were defined as the lowest and highest data points that were still within 1.5 of the interquartile range, excluding outliers (depicted as individual data points). Tmax time to reach maximal concentration, ODT orodispersible tablet
3.2.2 Pivotal Studies
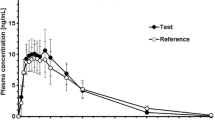
The mean plasma concentration versus time profiles of the 30 mg morphine sulfate ODT and the 3 × 10 mg tablets, and of the 2 × 5 mg morphine sulfate ODTs and the 10 mg tablet are depicted in Fig. 3. The profiles and calculated pharmacokinetic parameters for the ODTs and tablets were similar for both the 30 mg and 10 mg formulations (Table 2). The test-to-reference ratios of the geometric LS means and 90% CIs for the 30 mg formulations were 90.90% (84.86–97.36%) for the Cmax and 96.83% (94.35–99.37%) for the AUC0–t, and those for the 10 mg formulations were 96.13% (91.64–100.85%) for the Cmax and 99.88% (97.27–102.56%) for the AUC0–t (Table 3). All 90% CIs fell within the 80–125% range, confirming the bioequivalence of both doses of the ODT and tablet formulations under fasting conditions. Observed Tmax values were also similar for the 30 mg ODT and 3 × 10 mg tablets (0.78 h and 0.75 h, respectively), and for the 2 × 5 mg ODTs and the 10 mg tablet (0.76 h and 1.00 h, respectively) (Table 2).

Mean (SD) morphine plasma concentration versus time linear profiles following oral administration of a 30 mg of morphine sulfate as an ODT (1 × 30 mg, blue squares) or Sevredol® tablets (3 × 10 mg, green triangles) in healthy subjects (n = 80) under fasting conditions; and b 5 mg of morphine sulfate as an ODT (2 × 5 mg, blue squares) or Sevredol® tablet (1 × 10 mg, green triangles) in healthy subjects (n = 82) under fasting conditions in the pivotal studies. Inserts show the full kinetic profiles over 36 h. For graph readability, negative error bars were used for ODT data, and positive error bars were used for tablet data. ODT orodispersible tablet
Cmax and AUC were dose proportional between the 10 mg (2 × 5 mg ODTs) and 30 mg (1 × 30 mg ODT) doses. No dose-dependent changes in the Tmax (average: 0.8 h) or T½ (around 10–11 h) were observed for the ODTs.
3.3 Safety Analysis
Throughout all three studies, no SAEs were reported and no healthy subjects were withdrawn for safety reasons.
3.3.1 Pilot Study
Ninety-nine AEs were reported by 15/20 (75%) subjects. All of the AEs (87/99 [88%]) considered as related to one of the drugs (i.e., morphine or naltrexone) were of mild (66/87 [76%]) or moderate (21/87 [24%]) intensity. Their frequency was similar for the ODT (20/87 [23%]), capsule (14/87 [16%]) and tablet (20/87 [23%]) formulations, whereas drug-related AEs appeared to be slightly more frequent for the solution (33/87 [38%]). The most common drug-related AEs were neurological (somnolence, dizziness and headache) and gastrointestinal (nausea and vomiting) disorders, with similar profiles for the different formulations.
3.3.2 Pivotal Studies
In the 30 mg dose study, 63 AEs were reported by 23/42 (55%) subjects, and 45/63 (71%) were considered to be drug-related (morphine or naltrexone). For the 10 mg dose study, 67 AEs were reported by 22/42 (52%) subjects, and 47/67 (70%), reported by 15/42 [36%] subjects, were judged to be morphine-related. No naltrexone was administered. In both studies, the frequency of drug-related AEs was similar for the ODTs and tablets, with the most common events being the same neurological disorders reported in the pilot study. The drug-related AEs across both studies were generally of mild or moderate intensity but one severe AE (syncope) was reported with the 10 mg tablet 42 h after dosing and was resolved within 10 min.
3.4 Palatability
Among the subjects that evaluated palatability of the 30 mg ODT (17 subjects) and 30 mg/5 mL solution (19 subjects) during the pilot study, the taste/aroma was rated as neutral to very good by 35% for the ODT, compared with 16% for the oral solution (Fig. 4). Bitterness, formulation overall rating for oral administration and texture in the mouth were reported to be similar for the 30 mg ODT and the solution. Finally, treatment avoidance in case of intense pain due to palatability issues was considered unlikely by more than 75% of subjects for the ODT and the solution.

Product evaluation after oral administration of the orodispersible tablets (top blue bar) or solution (lower green bar) in the pilot study. ODT orodispersible tablet
4 Discussion
The results of the three randomized, laboratory-blinded, crossover, phase I studies, all conducted in healthy volunteers, provided the first comparative evaluations of the pharmacokinetic profile, safety, palatability and bioequivalence of a new ODT formulation of morphine sulfate, developed to facilitate the oral administration of pain relief in patients with swallowing difficulties. The pharmacokinetic profile, safety and bioavailability of the ODTs were shown to be similar to those of currently marketed oral morphine sulfate formulations across all three studies. Bioequivalence of 30 mg and 10 mg doses of the ODT with those of a reference tablet formulation was also demonstrated in two studies. In addition, the palatability of the ODTs was reported to be similar to that of a reference oral morphine solution.
In the pilot study, plasma exposure following the 30 mg morphine dose was found to be similar for all the oral formulations (including the ODT, tablet, capsule and oral solution) with AUC0–t differences below 10%. However, the 30 mg/5 mL solution was associated with a higher average Cmax. The range of Tmax values observed for the ODT (0.76–1.00 h) was similar to that observed for the tablet (0.75–1.00 h) across all three studies, and to the other reference formulations in the pilot study (0.75–1.25 h). The administration of all tested formulations was associated with an initial steep increase in plasma morphine concentrations; thus, the onset of action for morphine would be expected to be similar for all formulations. Oral immediate-release morphine formulations have generally been reported to take approximately 30 min to provide significant pain relief, due to the time needed for absorption through the gastrointestinal tract and the hepatic first-pass effect [13]. Pharmacokinetic/pharmacodynamic modeling has provided further insights into the time course of the clinical effects after opioid administration (extensively reviewed by Lötsch [14]), revealing that morphine has a slow plasma-effect-site transfer rate with an equilibrium half-life (T½,keo) ranging from 1.6 to 4.8 h. Thus, the slight variations in Tmax observed for the various different immediate-release formulations evaluated in the current studies would not be expected to influence the onset of efficacy of the drug, as the slow transfer of morphine to the effect site would be the rate-limiting step in achieving analgesia.
The safety profile of the ODTs was similar to those of the reference tablet, capsule and liquid formulations. Based on available data, the nature of the reported AEs was as expected following morphine administration, including the occurrence of syncope with the 10 mg tablet, which is listed as an uncommon event in the Summary of Product Characteristics (SmPC) for this formulation [15]. The assessment of the palatability of the ODTs during the pilot study did not identify any major issues with the texture of the ODT in the mouth, and overall findings indicated that the ODT had a similar palatability profile to that of the solution. However, it is important to note that this palatability assessment was carried out using the 30 mg ODT, and that this high-dose formulation contained the lowest ratio of flavor/sweetener to active substance of all the ODTs developed. Indeed, as all the ODTs across the range of doses were formulated to contain the same cumulated concentration of flavor and sweetener, the concentration of morphine sulfate modifies the bitterness of the formulations: the lower-dose ODTs are sweeter and therefore their taste would presumably be more acceptable for children, whereas the bitterness is more pronounced for the higher-dose ODTs intended for use in adult populations. Thus, although further studies are required, improved product appreciation would be expected among subjects administered the lower-dose ODT formulations containing higher flavor/sweetener to active substance ratios.
The few studies that have been conducted to date to evaluate the efficacy and safety of oral morphine in children aged 1–17 years to control pain at home or in hospital [16,17,18,19,20,21,22,23,24,25,26,27] used similar doses (100–500 µg/kg) to those recommended by the WHO guidelines for pain management (80–500 µg/kg) [28]. As for many drugs, achieving small doses with an oral solution formulation is difficult [29, 30]. ODT treatment in children has been reported to be appreciated by healthcare professionals, allowing for improved accuracy and easy administration [31], and to be well accepted by young children [32]. The range of morphine sulfate ODTs (1, 2.5, 5, 10, 20 and 30 mg) developed by Ethypharm enables coverage of all the recommended pediatric dosages, starting from infants aged 6 months, and allows titration to the lowest effective dose in each pediatric population as recommended by the guidelines [28].
The small size of the population in the pilot study prevented any definitive conclusions being drawn about the bioequivalence of the commercially available immediate-release formulations (tablet, capsules and solution) and the newly developed ODT. However, the results of this pilot study did demonstrate similarities between the tested formulations, and gave sufficient information about intrasubject variability to allow the size of the two pivotal study populations to be defined. Moreover, as the range of patients requiring an alternative to solid drug oral administration ranges from elderly to pediatric patients, it is important to highlight the potential of the ODTs to provide a convenient approach for morphine delivery. In addition to the rapid disintegration of the ODTs in the mouth, the ODTs can also be dispersed in a small volume of liquid in a spoon, most notably for facilitating administration in pediatric patients. The range of ODT morphine sulfate doses (1, 2.5, 5, 10, 20 and 30 mg) will also allow for accurate dosing of morphine according to patient body weight and age, and minimize the risk of dosing errors.
5 Conclusion
Oral administration of morphine sulfate in the form of ODTs resulted in similar safety and plasma exposure profiles as those observed for a range of reference oral formulations. Bioequivalence was also demonstrated between the 30 mg and 2 × 5 mg morphine sulfate ODTs and corresponding doses of a reference tablet (Sevredol® 10 mg) in two comparative pivotal bioavailability studies. These new morphine sulfate ODTs may therefore constitute an easy and accurate mode of oral morphine administration, enabling all patients with swallowing difficulties, ranging from pediatric to adult and elderly patients, to achieve and maintain relief from severe pain with the lowest possible morphine dose.
References
World Health Organization. WHO guidelines for the pharmacological and radiotherapeutic management of cancer pain in adults and adolescents. Geneva: World Health Organization; 2018. https://www.who.int/publications/i/item/9789241550390. Accessed 14 Jun 2022.
Martindale. Analgesics, anti-inflammatory drugs and antipyretics. Morphine. In: Brayfield A, editor. Martindale: the complete drug reference. 39th ed. London: Pharmaceutical Press; 2017. p. 93–7.
US FDA, Center for Drug Evaluation and Research. Morphine Sulfate Roxane Clinical pharmacology and biopharmaceutics reviews. Application number NDA 22-195 and NDA 22-207. Silver Spring, MD: US FDA; 2007.
Guo BB, Zhang YQ, Wang SF, Ding JS, Zhou WH. The pharmacokinetics of morphine and codeine in human plasma and urine after oral administration of Qiangli Pipa Syrup. J Forensic Sci. 2018;63(4):1221–8. https://doi.org/10.1111/1556-4029.13696.
Webster L, Owen J, Darling I, Richards P, Kelen R, Stern W (eds). Single-dose and steady-state pharmacokinetics of moxduo™, A dual-opioid formulation containing a fixed ratio of morphine and oxycodone [poster]. In: 29th Annual Meeting of the American Pain Society, 6–8 May 2010: Baltimore, MD.
World Health Organization. World Health Organization Model List of Essential Medicines for Children—8th list, 2021. WHO/MHP/HPS/EML/2021.03. Geneva: World Health Organization; 2021. https://www.who.int/publications/i/item/WHO-MHP-HPS-EML-2021.03. Accessed 14 Jun 2022.
World Health Organization. World Health Organization Model List of Essential Medicines, 22nd List, 2021. WHO/MHP/HPS/EML/2021.02. Geneva: World Health Organization; 2021. https://www.who.int/publications/i/item/WHO-MHP-HPS-EML-2021.02. Accessed 14 Jun 2022.
Fallon M, Giusti R, Aielli F, Hoskin P, Rolke R, Sharma M, et al. Management of cancer pain in adult patients: ESMO Clinical Practice Guidelines. Ann Oncol. 2018;29(Suppl 4):iv166–91. https://doi.org/10.1093/annonc/mdy152.
Dowell D, Haegerich TM, Chou R. CDC Guideline for Prescribing Opioids for Chronic Pain—United States, 2016. MMWR Recommendations and reports: morbidity and mortality weekly report recommendations and reports. Centers Dis Control Prev. 2016;65(1):1–49.
American Geriatrics Society. Panel on the pharmacological management of persistent pain in older persons. Pharmacological management of persistent pain in older persons. Pain Med. 2009;10(6):1062–83. https://doi.org/10.1111/j.1526-4637.2009.00699.x.
European Medicines Agency. Guideline on pharmaceutical development of medicines for paediatric use. EMA/CHMP/QWP/805880/2012 Rev. 2. London: European Medicines Agency; 2013. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-pharmaceutical-development-medicines-paediatric-use_en.pdf. Accessed 12 Jun 2022.
European Medicines Agency. Guideline on the investigation of bioequivalence. CPMP/EWP/QWP/1401/98 Rev. 1/ Corr **. London: European Medicines Agency; 20 Jan 2010. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-bioequivalence-rev1_en.pdf. Accessed 12 Jun 2022.
Coluzzi PH. Sublingual morphine: efficacy reviewed. J Pain Symptom Manag. 1998;16(3):184–92. https://doi.org/10.1016/s0885-3924(98)00046-3.
Lötsch J. Pharmacokinetic-pharmacodynamic modeling of opioids. J Pain Symptom Manag. 2005;29(5 Suppl):S90-103. https://doi.org/10.1016/j.jpainsymman.2005.01.012.
Napp Pharmaceuticals Limited. Summary of Product Characteristics. Sevredol tablets 10 mg. Last updated 4 Nov 2020. https://www.medicines.org.uk/emc/product/1020/smpc. Accessed 14 Jun 2022.
Wei Chern Gan R, Kamani T, Daniel M. Analgesia following adenotonsillar surgery in children: is Oramorph required in addition to paracetamol and ibuprofen? Eur Ann Otorhinolaryngol Head Neck Dis. 2017;134(1):23–5. https://doi.org/10.1016/j.anorl.2016.03.008.
Engelhardt T, Crawford M. Sublingual morphine may be a suitable alternative for pain control in children in the postoperative period. Paediatr Anaesth. 2001;11(1):81–3. https://doi.org/10.1046/j.1460-9592.2001.00598.x.
O’Hara M, McGrath PJ, D’Astous J, Vair CA. Oral morphine versus injected meperidine (Demerol) for pain relief in children after orthopedic surgery. J Pediatr Orthop. 1987;7(1):78–82. https://doi.org/10.1097/01241398-198701000-00016.
Syed MI, Magos TA, Singh J, Montague ML. A new analgesia regimen after (adeno) tonsillectomy in children: a pilot study. Clin Otolaryngol. 2016;41(6):660–5. https://doi.org/10.1111/coa.12579.
Dawes JM, Cooke EM, Hannam JA, Brand KA, Winton P, Jimenez-Mendez R, et al. Oral morphine dosing predictions based on single dose in healthy children undergoing surgery. Paediatr Anaesth. 2017;27(1):28–36. https://doi.org/10.1111/pan.13020.
Beale JP, Oglesby AJ, Jones A, Clancy J, Beattie TF. Comparison of oral and intravenous morphine following acute injury in children. Eur J Emerg Med. 2001;8(4):271–4. https://doi.org/10.1097/00063110-200112000-00004.
Kelly LE, Sommer DD, Ramakrishna J, Hoffbauer S, Arbab-Tafti S, Reid D, et al. Morphine or Ibuprofen for post-tonsillectomy analgesia: a randomized trial. Pediatrics. 2015;135(2):307–13. https://doi.org/10.1542/peds.2014-1906.
Delgove A, Harper L, Berciaud S, Lalioui A, Angelliaume A, Lefevre Y. Efficacy, pain, and overall patient satisfaction with pediatric upper arm fracture reduction in the emergency department. Orthop Traumatol Surg Res. 2019;105(3):513–5. https://doi.org/10.1016/j.otsr.2018.10.027.
Chasle V, de Giorgis T, Guitteny MA, Desgranges M, Metreau Z, Herve T, et al. Evaluation of an oral analgesia protocol for upper-limb fracture reduction in the paediatric emergency department: Prospective study of 101 patients. Orthop Traumatol Surg Res. 2019;105(6):1199–204. https://doi.org/10.1016/j.otsr.2019.06.009.
Poonai N, Bhullar G, Lin K, Papini A, Mainprize D, Howard J, et al. Oral administration of morphine versus ibuprofen to manage postfracture pain in children: a randomized trial. Can Med Assoc J. 2014;186(18):1358–63. https://doi.org/10.1503/cmaj.140907.
Poonai N, Datoo N, Ali S, Cashin M, Drendel AL, Zhu R, et al. Oral morphine versus ibuprofen administered at home for postoperative orthopedic pain in children: a randomized controlled trial. Can Med Assoc J. 2017;189(40):E1252–8. https://doi.org/10.1503/cmaj.170017.
Wille C, Bocquet N, Cojocaru B, Leis A, Chéron G. Oral morphine administration for children’s traumatic pain [in French]. Arch Pediatr. 2005;12(3):248–53. https://doi.org/10.1016/j.arcped.2004.07.021.
World Health Organization. Persisting pain in children package: WHO guidelines on the pharmacological treatment of persisting pain in children with medical illnesses. Geneva: World Health Organization; 2012. https://apps.who.int/iris/handle/10665/44540. Accessed 30 Sep 2022.
Comoglu T, Dilek OE. Orally disintegrating tablets and orally disintegrating mini tablets—novel dosage forms for pediatric use. Pharm Dev Technol. 2019;24(7):902–14. https://doi.org/10.1080/10837450.2019.1615090.
Lopez FL, Ernest TB, Tuleu C, Gul MO. Formulation approaches to pediatric oral drug delivery: benefits and limitations of current platforms. Expert Opin Drug Deliv. 2015;12(11):1727–40. https://doi.org/10.1517/17425247.2015.1060218.
Rautamo M, Kvarnström K, Sivén M, Airaksinen M, Lahdenne P, Sandler N. A focus group study about oral drug administration practices at hospital wards-aspects to consider in drug development of age-appropriate formulations for children. Pharmaceutics. 2020;12(2):109. https://doi.org/10.3390/pharmaceutics12020109.
Slavkova M, Breitkreutz J. Orodispersible drug formulations for children and elderly. Eur J Pharm Sci. 2015;75:2–9. https://doi.org/10.1016/j.ejps.2015.02.015.
Acknowledgments
Medical writing assistance was provided by Drs Céline Zimmer, Emma Pilling and Marielle Romet (Santé Active Edition) and was funded by Ethypharm. Clinical studies, bioanalyses and pharmacokinetic analyses were accomplished by Algorithme Pharma, now Altasciences (Quebec, Canada) and were funded by Ethypharm.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was funded by Ethypharm SAS.
Conflicts of Interest
Nicolas Atrux-Tallau, Zulaikha Naimi, and Eric-Olivier Jaudinot are employed by Ethypharm SAS.
Ethics Approval
All studies were conducted in accordance with the ethical principles of the Declaration of Helsinki, the ICH Guideline E6 for GCP, European regulation EU 536/2014, and the Tri-Council Policy Statement (Canada). The study protocols were approved by Health Canada and by an independent Institutional Review Board (Advarra).
Consent to Participate
Informed consent was obtained from all participants included in this study.
Consent to Publish
All data were anonymized and participants were informed that the results of this study may be subject to publication and presentation in meetings.
Data Availability
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
Code availability
Not applicable.
Author Contributions
Material preparation, data collection and analysis were performed by NAT, ZN and EOJ. The first draft of the manuscript was written by NAT and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Atrux-Tallau, N., Naimi, Z. & Jaudinot, EO. Pharmacokinetics of Morphine Sulfate Orodispersible Tablets and Bioequivalence with Immediate-Release Oral Morphine Sulfate Formulations in Healthy Adult Subjects Under Fasting Conditions: Single-Dose Comparative Bioavailability Studies. Clin Drug Investig 42, 1101–1112 (2022). https://doi.org/10.1007/s40261-022-01214-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40261-022-01214-x