
Abstract
Background and Objectives
This study examined the effects of moderate renal impairment on the pharmacokinetics and pharmacodynamics of canagliflozin in Japanese patients with type 2 diabetes mellitus.
Methods
Japanese patients with stable type 2 diabetes (12 with moderate renal impairment and 12 with normal renal function or mild renal impairment) were eligible. This was an open-label, randomized, two-way crossover, two-sequence, single-dose study performed at a single center in Japan. The subjects were hospitalized for the pharmacodynamic/pharmacokinetic evaluations. Twenty-four patients received a single dose each of canagliflozin 100 and 200 mg before breakfast in a crossover manner with a 14-day washout between doses. The main outcome measures were pharmacokinetics of canagliflozin and its main metabolites (M5 and M7) in plasma and urine, and change from baseline in 24-h urinary glucose excretion (ΔUGE24 h).
Results
There was no significant effect of moderate renal impairment on the maximum canagliflozin concentration. The ratios of least square means (90 % confidence intervals [CIs]) of moderate renal impairment relative to normal renal function or mild renal impairment were 0.982 (0.821–1.173) and 0.989 (0.827–1.182) for the 100 and 200 mg doses, respectively. The canagliflozin area under the plasma concentration–time curve was greater in those with moderate renal impairment than in those without, after both canagliflozin doses (ratio of least square means [90 % CI] 1.258 [1.061–1.490] and 1.216 [1.026–1.441]). ΔUGE24 h increased after administration of both doses, but in patients with moderate renal impairment, the increase was approximately 70 % of that in patients with normal renal function or mild renal impairment. The incidence of adverse events was low and no patient developed hypoglycemia.
Conclusion
The pharmacokinetics of canagliflozin are affected by renal function, with slight decreases in renal clearance observed. No effect of renal impairment on the maximum concentration was observed. Renal impairment reduced the ability of canagliflozin to promote urinary glucose excretion.
Similar content being viewed by others

Canagliflozin pharmacokinetics are unknown in Japanese patients with type 2 diabetes mellitus and chronic kidney disease. |
Moderate renal impairment increased the canagliflozin area under the plasma concentration–time curve but not the maximum concentration in Japanese patients with type 2 diabetes, and somewhat attenuated the increase in urinary glucose excretion that occurs with canagliflozin treatment in patients with normal renal function or mild renal impairment. |
Canagliflozin may be a suitable treatment option for Japanese patients with type 2 diabetes and moderate renal impairment. |
1 Introduction
Chronic kidney disease (CKD) occurs in 20–30 % of patients with type 2 diabetes mellitus globally, according to several reports [1–3]. One US study reported CKD prevalence to be as high as 39.6 % among people with diagnosed diabetes and 41.7 % in those with undiagnosed diabetes [4]. The prevalence of CKD in the general Japanese population is estimated at 13 % [5]. Studies from the USA show that the prevalence of CKD is increasing over time. De Boer et al reported that the prevalence of diabetic kidney disease increased from 2.2 % of the US population in 1988 to 3.3 % in 2008 (p < 0.001) in proportion to the reported increase in the prevalence of diabetes [6], while Kramer and Molitch [7] reported a fourfold increase in prevalence of CKD in the last three decades in US Medicare patients with hypertension and diabetes. However, CKD often goes undetected, so the actual prevalence of the disease may be even higher than reported.
Treatment options for patients with type 2 diabetes and CKD are limited. Glucose-lowering agents need to be dose-adjusted and are often contraindicated in patients with stage 3 CKD or higher because of the risk of hypoglycemia [8, 9]. Many, including sulfonylureas, glinides, and thiazolidinediones, also cause weight gain, while the only weight-neutral agent, metformin, is contraindicated or its use is limited in this population [1]. Further treatment options are clearly required.
Canagliflozin is an inhibitor of sodium glucose co-transporter 2 (SGLT2), which is expressed predominantly in the proximal renal tubules, and is responsible for the majority of glucose reabsorption from urine [10]. SGLT2 inhibitors act independently of insulin. The main canagliflozin-metabolizing enzyme is uridine diphosphate (UDP)-glucuronosyltransferase (UGT), and the main metabolites of canagliflozin are the O-glucuronides M5 and M7, both of which are inactive (FDA Endocrinologic and Metabolic Drugs Advisory Committee, NDA 204042, 10 Jan 2013, unpublished data).
By inhibiting SGLT2 expressed in the proximal renal tubules, canagliflozin reduces reabsorption of filtered glucose and lowers the renal threshold for glucose, thereby increasing urinary glucose excretion (UGE) and resulting in a net caloric loss, which may help with weight loss [11–15]. Inagaki et al. [16] found that canagliflozin significantly reduced body weight in Japanese patients with type 2 diabetes. However, because UGE is related to the glomerular filtration rate (GFR), the action of canagliflozin to increase UGE may be affected by decreased renal function [12, 13, 17].
In non-Japanese patients with type 2 diabetes, canagliflozin has shown promising glucose-lowering effects in those with [18] or without [19] kidney disease, and its pharmacokinetics appear to support once-daily dosing [20]. However, the pharmacokinetic profile of canagliflozin has not been assessed in Japanese patients with type 2 diabetes and impaired renal function. Therefore, the primary objective of this study was to compare the pharmacokinetic profiles of canagliflozin and its main metabolites M5 and M7 in Japanese patients with type 2 diabetes with or without moderate renal impairment after a single dose of canagliflozin 100 or 200 mg. Secondary objectives were to assess the pharmacodynamic effects and safety of canagliflozin in the different groups.
2 Methods
2.1 Study Design
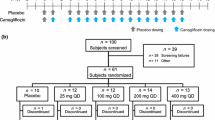
The study followed an open-label, randomized, two-way crossover, single-dose design. Patients with moderate renal impairment, mild renal impairment, or normal renal function were randomized to one of two treatment sequences (n = 6 per sequence), and each patient received a single dose of canagliflozin 100 or 200 mg 10 min before breakfast. After a washout period of at least 14 days, a second dose (opposite to the first) was administered. Follow-up examinations were performed 14–21 days after the second treatment administration. Randomization was achieved using a list of randomization key codes, and the treatment sequence was allocated on the basis of an ascending order of key codes by a study registration center.
The study protocol and all amendments were approved by the institutional review board of the study site (P-One Clinic, Keikokai Medical Corporation, Tokyo, Japan), and the study was conducted in accordance with the ethical principles of the Declaration of Helsinki, the Pharmaceutical Affairs Law, and Good Clinical Practice. All patients provided written informed consent.
If a randomized subject was removed from the study before study drug administration in Period I, the subject was to be replaced by another subject. At the time of the subject’s discontinuation, the investigator selected a replacement subject who was assigned the smallest subject identification code among those who were assessed as eligible based on the screening examination and not randomized, and were assigned to the treatment group that was originally assigned to the discontinued subject. If there were no eligible subjects at the time of discontinuation, the principal investigator or sub-investigators were to select a replacement subject with the smallest subject identification code among those who were assessed as eligible based on screening examinations conducted after discontinuation. To facilitate recruitment and replacement, the protocol was amended to allow the prospective enrolment of eligible subjects who could ‘stand by’ to serve as replacements, as needed, for subjects who withdrew before study drug administration; if the subject was replaced by a standby subject, the standby subject was assigned the same identification number as the discontinued subject. The purpose of this amendment was to facilitate recruitment and subject replacement.
The subjects were hospitalized for the pharmacodynamic/pharmacokinetic evaluations, and their meals (breakfast, lunch, and dinner) were provided by the study site during this time. The intake of other food was prohibited during hospitalization, except for glucose in the event of hypoglycemia. The meal contents were identical in both treatment periods. Breakfast, lunch, and dinner were served at 10 min, 4 h 30 min, and 10 h 30 min after the scheduled study drug administration, and all meals were to be consumed within 20 min of serving.
2.2 Subjects
Twenty-four patients aged 40–79 years with stable type 2 diabetes (12 with moderate renal impairment and 12 with normal renal function or mild renal impairment) were initially enrolled and completed the study. The eligibility criteria are listed in Table 1. Patients using oral drugs entered a 14-day washout after providing informed consent and before the screening visit.
2.3 Plasma Pharmacokinetic Parameters
Blood samples for measurement of total (free and bound) canagliflozin and its metabolites concentrations in plasma were taken a total of 13 times in each dosing period: immediately before the dose, and 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, 48, and 72 h post-dose. Blood was drawn in K2EDTA blood collection tubes, cooled in iced water, and centrifuged. The obtained plasma was frozen immediately and stored at −20 °C until analysis. The plasma concentrations of unchanged canagliflozin were measured by high-performance liquid chromatography (HPLC)/tandem mass spectrometry (HPLC–MS/MS; HPLC unit: Alliance® 2795 separations module, Waters, Milford, MA, USA; mass spectrometry unit: API4000™, AB SCIEX, Framingham, MA, USA; L-column ODS, 2.1 × 50 mm, particle size 5 μM, Chemicals Evaluation and Research Institute, Tokyo, Japan) with 13C6-canagliflozin as an internal standard. Plasma samples were loaded into an OASIS® HLB (Waters, Milford, MA, USA) and eluted with methanol. The eluate was evaporated to dryness under a stream of nitrogen gas. The residue was dissolved and injected into the LC-MS/MS system. The validated quantification range was 1–2,000 ng/mL. All validation results satisfied the predefined acceptance criteria. The plasma concentrations of metabolites (M5 and M7) were measured using a previously described method [20]. The method used to prepare samples for the determination of metabolites (M5 and M7) involved protein precipitation with acetonitrile followed by HPLC-MS/MS. The validated quantification range for the metabolites (M5 and M7) was 5–10,000 ng/mL [20].
2.4 Urinary Pharmacokinetic Parameters
Seven urine samples were taken in each treatment period: 11–0 h pre-dose on the preceding day, 0–4.5, 4.5–10.5, 10.5–13, and 13–24 h post-dose on administration day, then 24–48 and 48–72 h post-dose. The volume of urine at each timepoint was calculated and described in a case report form. After the volume was calculated, urine was mixed and transferred to two polypropylene tubes (total 5 mL) and immediately stored at −20 °C. Samples were stored at −20 °C until analysis. The urinary canagliflozin concentrations were determined by solid-phase extraction of urine samples followed by HPLC–MS/MS, which was performed as described for the determination of plasma canagliflozin. The validated quantification range was 1–2,000 ng/mL. All validation results satisfied the predefined acceptance criteria.
2.5 Pharmacodynamic Evaluations
Urinary glucose concentrations and excretion rates over a 24-h period were assessed. Ten samples were taken in each treatment period, at 0–4.5, 4.5–10.5, 10.5–13, and 13–24 h after the scheduled dose on the day preceding administration of the study drug, 0–4.5, 4.5–10.5, 10.5–13, and 13–24 h post-dose on administration day, and 24–48 and 48–72 h post-dose.
Plasma glucose concentrations were measured in 33 blood samples obtained in each patient, as follows. Sixteen samples were taken on the day preceding administration: at the time of the scheduled dose (before breakfast), and 0.5, 1, 1.5, 2, 3, 4.5 (immediately before lunch), 5.5, 6, 7, 8, 10.5 (immediately before dinner), 11.5, 12, 13, and 14 h after the scheduled dose. Sixteen samples were taken on the administration day: immediately before the dose, and 0.5, 1, 1.5, 2, 3, 4.5 (immediately before lunch), 5.5, 6, 7, 8, 10.5 (immediately before dinner), 11.5, 12, 13, and 14 h post-dose. Finally, one additional sample was taken the next day (24 h post-dose, before breakfast). Glucose concentrations were also measured in urine samples taken immediately before the dose and at 24 h post-dose (before breakfast).
2.6 Safety
Adverse events were recorded along with information on their intensity and relationship to the study drug. Adverse events were classified according to system organ class and preferred term [MedDRA® (the Medical Dictionary for Regulatory Activities)/J version 15.0]. Adverse drug reactions were defined as events that were considered reasonably related to the study drug by the investigator. Other safety assessments included blood and urine laboratory parameters, renal function, vital signs (blood pressure, pulse rate, body temperature), body weight, and 12-lead electrocardiogram.
2.7 Statistical Methods
Statistical methods are described in the online Electronic Supplementary Material.
3 Results
3.1 Patient Characteristics
All 24 patients received at least one dose of canagliflozin and completed the trial (Electronic Supplementary Material Fig. 1). There were some minor protocol violations: ten subjects took longer than 20 min to finish a meal; two subjects did not eat an entire meal; and one subject had a hemolyzed blood sample so some laboratory parameters were classified as missing in this subject (namely, aspartate aminotransferase, alkaline phosphatase, lactate dehydrogenase, total protein, potassium, calcium, and inorganic phosphorus). None of these violations prevented any of the patients from being included in the analyses. No subject replacements were necessary. The characteristics of patients are summarized in Table 2.
3.2 Pharmacokinetics of Canagliflozin
The plasma concentrations of unchanged canagliflozin and its metabolites in patients with and without moderate renal impairment are shown in Fig. 1, and key pharmacokinetic parameters for each group are shown in Table 3. Canagliflozin was rapidly absorbed, reaching its maximum concentration (C max) within a median of 1 h. The half-life (t ½) in patients with normal or mild renal impairment was just over 12 h at both doses. The area under the plasma concentration–time curve (AUC) increased in a dose-dependent manner. Renal clearance (CLR) was low and the amount of canagliflozin excreted into urine from 0 to 72 h was <1 % of the dose for both doses.

Mean plasma concentration–time profiles for a canagliflozin, b M5, and c M7. Error bars show standard deviation. For clarity, only the upper error bars are shown
No difference in C max was seen between those with moderate renal impairment and those with normal renal function or mild renal impairment. The ratio of least square means (90 % confidence interval [CI]) of moderate renal impairment relative to normal renal function or mild renal impairment was estimated to be 0.982 (0.821–1.173) and 0.989 (0.827–1.182) for the 100 and 200 mg doses, respectively. However, AUC was greater in those with moderate renal impairment than in those with normal renal function or mild renal impairment following both doses of canagliflozin. The ratio of least square means (90 % CI) of moderate renal impairment relative to normal renal function or mild renal impairment was estimated at 1.258 (1.061–1.490) for the 100 mg dose and 1.216 (1.026–1.441) for the 200 mg dose. Analysis of variance showed significant differences between doses for C max and AUC (p < 0.001). There was no significant difference between patients with moderate renal impairment and those with normal renal function or mild renal impairment for C max, whereas the difference was significant for AUC (p = 0.039). The t ½ was slightly prolonged. CLR was slightly lower in patients with moderate renal impairment, but the amount of canagliflozin excreted into urine was unchanged relative to those in patients with normal renal function or mild renal impairment.
3.3 Pharmacokinetics of M5 and M7
The C max of both M5 and M7 occurred 2 h post-exposure at both doses, indicating that both metabolites were rapidly produced after administration of canagliflozin. Like canagliflozin, exposure in terms of AUC was dose dependent. Their t ½ values were also similar to that of canagliflozin. No difference in the C max of M5 was seen between patients with moderate renal impairment and those with normal renal function or mild renal impairment, but AUC was greater in those with renal impairment than in those without. The ratios of least square means (90 % CI) of moderate renal impairment relative to normal renal function or mild renal impairment for the C max and AUC of M5 were estimated to be 1.148 (0.945–1.395) and 1.655 (1.312–2.087), respectively, for 100 mg, and 1.218 (1.002–1.479) and 1.639 (1.299–2.067), respectively, for 200 mg. There were significant differences between doses for C max and AUC (p < 0.001). There was no significant difference between patients with moderate renal impairment and those with normal renal function or mild renal impairment for C max, whereas the difference between patient groups was significant for AUC (p = 0.001). The M5 t ½ was slightly prolonged in patients with impaired renal function compared with those with normal renal function or mild renal impairment, after both doses.
For M7, both C max and AUC were greater in patients with moderate renal impairment than in patients with normal renal function or mild renal impairment. The ratios of least square means (90 % CI) of moderate renal impairment relative to normal renal function or mild renal impairment for the C max and AUC of M7 were estimated to be 1.306 (1.064–1.603) and 1.709 (1.375–2.124) for 100 mg, and 1.342 (1.093–1.646) and 1.669 (1.343–2.074) for 200 mg. There were significant differences between doses for C max and AUC (p < 0.001), and the difference between the two patient groups was also significant for C max and AUC (p = 0.023 and p < 0.001, respectively). The t ½ of M7 was slightly prolonged in patients with moderate renal impairment.
3.4 Pharmacodynamics of Canagliflozin
The mean change from baseline in 24 h UGE (ΔUGE24 h) in patients with and without moderate renal impairment is shown in Fig. 2. ΔUGE24 h increased after administration of both doses of canagliflozin in both patient groups. However, ΔUGE24 h in patients with moderate renal impairment was approximately 70 % of that observed in patients with normal renal function or mild renal impairment.

Mean changes in cumulative 24-h urinary glucose excretion. Error bars show standard deviation. UGE urinary glucose excretion
The 24-h mean percent inhibition of renal glucose reabsorption after administration of canagliflozin was slightly higher in patients with moderate renal impairment than in those with normal renal function or mild renal impairment (Fig. 3).

Mean percent inhibition of renal glucose reabsorption over 24 h. Error bars show standard deviation
The plasma glucose concentrations after administration of canagliflozin decreased at almost all measurement timepoints in both patients with normal renal function or mild renal impairment and those with moderate renal impairment. In addition, the decrease in plasma glucose concentrations was smaller in patients with moderate renal impairment than in those with normal renal function or mild renal impairment.
The 24-h mean plasma glucose concentrations after administration of canagliflozin decreased in both patients with normal renal function or mild renal impairment and in patients with moderate renal impairment (Fig. 4). As for plasma glucose, the decrease in 24-h mean plasma glucose concentrations was smaller in patients with moderate renal impairment than in patients with normal renal function or mild renal impairment.

Changes in 24-h mean plasma glucose concentrations. Error bars show standard deviation. MPG mean plasma glucose
3.5 Safety
Table 4 presents a summary of adverse events by preferred term in patients with and without moderate renal impairment following single doses of canagliflozin 100 and 200 mg. There were no adverse events leading to study discontinuation, deaths, other serious adverse events or other significant adverse events. Two adverse events were assessed as moderate in intensity (one event of constipation after canagliflozin 100 mg and one of nasopharyngitis after 200 mg, both in patients with normal renal function or mild renal impairment). The remaining nine events were assessed as mild. Only one case of pollakiuria and one of diarrhea were considered to be related to the study drug. Both events were mild and resolved. The incidence of adverse events was not affected by renal function or canagliflozin dose. None of the patients developed hypoglycemia.
4 Discussion
This study assessed the pharmacokinetics of two doses of canagliflozin in Japanese patients with type 2 diabetes and moderate renal impairment or normal renal function/mild renal impairment. Canagliflozin was rapidly absorbed, with a relatively long t ½. Importantly, UGE continued to increase from day 2 onward, suggesting that a once-daily dosing regimen may be possible. The metabolites M5 and M7 were formed rapidly after administration, and their t ½ values were similar to the t ½ of unchanged canagliflozin.
The pharmacokinetics of canagliflozin in Japanese patients with type 2 diabetes and normal renal function or mild renal impairment were similar to that reported in Western patients. Devineni et al. [20] administered canagliflozin 50, 100, or 300 mg to patients with type 2 diabetes with normal or mildly decreased renal function for 7 days, and evaluated the pharmacokinetics after single and multiple doses. After a single dose of 100 mg, the C max was 1,096 ng/mL in the Western population [20] compared with 1,214 ng/mL in the Japanese population with mild or no renal impairment in the current study. The corresponding values for the canagliflozin AUC from time zero to 24 h were 6,357 and 5,645 ng·h/mL. The canagliflozin AUC and C max increased dose dependently in the present study and in the study by Devineni et al. [20].
Although decreased clearance of canagliflozin was observed in patients with renal impairment, the urinary excretion ratio of unchanged drug was <1 % in our study. This difference in canagliflozin clearance is not likely to be due to altered protein binding in patients with renal impairment, because total protein and albumin levels were similar between those with moderate renal impairment and those with normal renal function/mild renal impairment (data not shown). In non-Japanese patients with normal renal function or mild renal impairment, the mean 24-h urinary excretion ratios for M5 and M7 following a single dose of canagliflozin 100 mg were 8.11 and 25.1 %, respectively, both of which are higher than the ratio for unchanged canagliflozin (0.55 %) [20]. Accordingly, the observed increases in the plasma concentrations of M5 and M7 in our study can be partly explained by lowered CLR of the metabolites resulting from reduced renal function.
Other SGLT2 inhibitors have yielded similar results in patients with renal impairment. Macha et al. [21] reported decreased CLR and moderately increased systemic exposure to empagliflozin in patients with moderate renal impairment, while Kasichayanula et al. [22] found that the C max and AUC for dapagliflozin increased incrementally with declining renal function.
Although canagliflozin exposure was slightly increased as a result of decreased renal function, ΔUGE24 h was lower in patients with moderate renal impairment than in those with normal renal function or mild renal impairment. Despite the reduction in ΔUGE24 h as a result of decreased renal function, the percent inhibition of renal glucose reabsorption was slightly higher in patients with impaired renal function than in those with normal renal function or mild renal impairment. This may have resulted from a difference in SGLT2 expression in patients with reduced renal function. Reduced expression of SGLT2 has been observed in the remnant kidney of five of six nephrectomized rats, a model of chronic renal failure [23], and it is therefore possible that SGLT2 expression may also be slightly decreased in patients with moderate renal impairment. Another possibility is that there was a difference in canagliflozin exposure in the active site in these patients. However, because there was no difference in the cumulative urinary excretion of canagliflozin among the three groups of patients with different levels of renal function, it is unlikely that there was a difference in canagliflozin exposure in the renal tubules. The exact reason for the discrepancy therefore remains unclear.
Other SGLT2 inhibitors have also been shown to increase UGE in patients with impaired renal function. Ipragliflozin significantly increased UGE in each estimated GFR (eGFR) class in a study by Ferrannini et al. [24]. In this same study, absolute glycosuria decreased with declining GFR, while fractional glucose excretion (excretion/filtration; a measure of the efficiency of SGLT2 inhibitors) was unaffected by renal impairment. Because UGE is affected by the rate of glomerular filtration of glucose, the decrease in ΔUGE24 h may result from decreased renal function.
In type 2 diabetes patients with moderate renal impairment, the blood glucose-lowering effect is likely to be attenuated associated with decreased ΔUGE24 h. However, in these patients, the 24-h UGE was approximately 70 % of that in type 2 diabetes patients with normal renal function or mild renal impairment (60–70 g), and a decrease in 24-h mean plasma glucose was also observed. Based on this observation, we expect canagliflozin to reduce blood glucose levels in Japanese patients with moderate renal impairment. In addition, a phase III study in non-Japanese type 2 diabetes patients with moderate renal impairment (eGFR ≥30 and <50 mL/min/1.73 m2) demonstrated a significant blood glucose-lowering effect of canagliflozin in these patients compared with placebo [18].
Both doses of canagliflozin appeared to be well-tolerated: only one case of pollakiuria and one of diarrhea were considered related to the study drug. Both were mild and resolved. There were no cases of hypoglycemia. However, as this was a pharmacokinetic study in which only one dose was administered, the safety of canagliflozin in patients with renal impairment should be examined in larger, longer-term studies.
The results of this study should be considered in light of its limitations, including its small sample size and enrolment of Japanese patients. Therefore, the results may not be generalizable to other populations. However, our findings were consistent with those in Western patients reported by Devineni et al. [20]. In addition, we only studied patients with moderate renal impairment, so the findings may be different in patients with more severe kidney disease. Finally, we only administered single doses so we cannot infer whether the pharmacokinetics of canagliflozin and its metabolites would be affected during longer-term administration. Longer-term studies in a larger number of patients are needed to clarify this issue.
5 Conclusion
The pharmacokinetics of canagliflozin and its metabolites (M5 and M7) in Japanese patients are affected by renal function, with slight decreases in CLR found. No difference in the C max of canagliflozin was observed, but the C max of M5 and M7 was greater in patients with normal renal function or mild renal impairment than in patients with moderate renal impairment. In patients with moderately decreased renal function, the exposure to canagliflozin is greater than in those with normal renal function or mild renal impairment, although the increases in exposure were minimal. The ability of canagliflozin to promote UGE was reduced as a function of decreasing GFR. In Japanese type 2 diabetes patients with and without moderate renal impairment, canagliflozin administered as a single dose was well-tolerated, raising no safety concerns. The small increase in exposure to canagliflozin in patients with moderately decreased renal function is unlikely to be clinically relevant; thus, dose reductions may not be necessary and canagliflozin may be a suitable treatment option for Japanese patients with type 2 diabetes and moderate renal impairment.
References
Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes: a patient-centered approach. Position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care. 2012;35:1364–79.
Oulahiane A, Anaddam S, Ouleghzal H, et al. Diabetes management issues for patients with chronic kidney disease [in French]. Nephrol Ther. 2012;8:135–40.
Travers K, Martin A, Khankhel Z, et al. Burden and management of chronic kidney disease in Japan: systematic review of the literature. Int J Nephrol Renovasc Dis. 2013;6:1–13.
Plantinga LC, Crews DC, Coresh J, et al. Prevalence of chronic kidney disease in US adults with undiagnosed diabetes or prediabetes. Clin J Am Soc Nephrol. 2010;5:673–82.
Iimori S, Noda Y, Okado T, et al. Baseline characteristics and prevalence of cardiovascular disease in newly visiting or referred chronic kidney disease patients to nephrology centers in Japan: a prospective cohort study. BMC Nephrol. 2013;14:152.
de Boer IH, Rue TC, Hall YN, et al. Temporal trends in the prevalence of diabetic kidney disease in the United States. JAMA. 2011;305:2532–9.
Kramer H, Molitch ME. Screening for kidney disease in adults with diabetes. Diabetes Care. 2005;28:1813–6.
Flynn C, Bakris G. Noninsulin glucose-lowering agents for the treatment of patients on dialysis. Nat Rev Nephrol. 2013;9:147–53.
Haneda M, Morikawa A. Which hypoglycaemic agents to use in type 2 diabetic subjects with CKD and how? Nephrol Dial Transplant. 2009;24:338–41.
Nair S, Wilding JP. Sodium glucose cotransporter 2 inhibitors as a new treatment for diabetes mellitus. J Clin Endocrinol Metab. 2010;95:34–42.
Nomura S, Sakamaki S, Hongu M, et al. Discovery of canagliflozin, a novel C-glucoside with thiophene ring, as sodium-dependent glucose cotransporter 2 inhibitor for the treatment of type 2 diabetes mellitus. J Med Chem. 2010;53:6355–6.
Sha S, Devineni D, Ghosh A, et al. Canagliflozin, a novel inhibitor of sodium glucose co-transporter 2, dose dependently reduces calculated renal threshold for glucose excretion and increases urinary glucose excretion in healthy subjects. Diabetes Obes Metab. 2011;13:669–72.
Liang Y, Arakawa K, Ueta K, et al. Effect of canagliflozin on renal threshold for glucose, glycemia, and body weight in normal and diabetic animal models. PLoS One. 2012;7:e30555.
Rosenstock J, Aggarwal N, Polidori D, et al. Dose-ranging effects of canagliflozin, a sodium-glucose cotransporter 2 inhibitor, as add-on to metformin in subjects with type 2 diabetes. Diabetes Care. 2012;35:1232–8.
Devineni D, Morrow L, Hompesch M, et al. Canagliflozin improves glycemic control over 28 days in subjects with type 2 diabetes not optimally controlled on insulin. Diabetes Obes Metab. 2012;14:539–45.
Inagaki N, Kondo K, Yoshinari T, et al. Efficacy and safety of canagliflozin in Japanese patients with type 2 diabetes: a randomized, double-blind, placebo-controlled, 12-week study. Diabetes Obes Metab. 2013;15:1136–45.
Polidori D, Sakai M, Devineni D. Exposure-response modeling of canagliflozin effects on the renal glucose threshold in subjects with type 2 diabetes (T2DM) [abstract no. 1072-P]. Diabetes. 2011; 60(Suppl. 1):A294.
Yale JF, Bakris G, Cariou B, et al. Efficacy and safety of canagliflozin in subjects with type 2 diabetes and chronic kidney disease. Diabetes Obes Metab. 2013;15:463–73.
Cefalu WT, Leiter LA, Yoon KH, et al. Efficacy and safety of canagliflozin versus glimepiride in patients with type 2 diabetes inadequately controlled with metformin (CANTATA-SU): 52 week results from a randomised, double-blind, phase 3 non-inferiority trial. Lancet. 2013;382:941–50.
Devineni D, Curtin CR, Polidori D, et al. Pharmacokinetics and pharmacodynamics of canagliflozin, a sodium glucose co-transporter 2 inhibitor, in subjects with type 2 diabetes mellitus. J Clin Pharmacol. 2013;53:601–10.
Macha S, Mattheus M, Halabi A, et al. Pharmacokinetics, pharmacodynamics and safety of empagliflozin, a sodium glucose cotransporter 2 (SGLT2) inhibitor, in subjects with renal impairment. Diabetes Obes Metab. 2014;16:215–22.
Kasichayanula S, Liu X, Benito MP, et al. The influence of kidney function on dapagliflozin exposure, metabolism, and efficacy in healthy subjects and in patients with type 2 diabetes mellitus. Br J Clin Pharmacol. 2012;76:732–44.
Nakamura N, Masuda S, Takahashi K, et al. Decreased expression of glucose and peptide transporters in rat remnant kidney. Drug Metab Pharmacokinet. 2004;19:41–7.
Ferrannini E, Veltkamp SA, Smulders RA, et al. Renal glucose handling: impact of chronic kidney disease and sodium-glucose cotransporter 2 inhibition in patients with type 2 diabetes. Diabetes Care. 2013;36:1260–5.
Matsuo S, Imai E, Horio M, et al. Collaborators developing the Japanese equation for estimated GFR. Revised equations for estimated GFR from serum creatinine in Japan. Am J Kidney Dis. 2009;53:982–92.
Acknowledgments
Canagliflozin is being developed by Mitsubishi Tanabe Pharma Corporation in collaboration with Janssen Research & Development, LLC. This study was funded by Mitsubishi Tanabe Pharma Corporation. The authors wish to acknowledge Dr. Nicholas D. Smith, Ph.D., and Ms. Marion Barnett for providing medical writing support during the preparation of this manuscript.
Disclosures
N.I. has received consulting fees, payment for reviewing the manuscript and research support from, and is a speaker for, Mitsubishi Tanabe Pharma Corporation. N.I. has also received consulting fees and/or research support from Taisho Pharmaceutical Co., Ltd., Takeda Pharmaceutical Company Ltd., Nippon Boehringer Ingelheim Co., Ltd., GlaxoSmithKline K.K., MSD K.K., Eli Lilly Japan K.K., Novartis Pharma K.K., Sanofi K.K., Daiichi Sankyo Company, Ltd., Astellas Pharma Inc., AstraZeneca K.K., and Bristol-Myers K.K., and is a speaker for MSD K.K., Novartis Pharma K.K., Sanofi K.K., Nippon Boehringer Ingelheim Co., Ltd., Ono Pharmaceutical Co., Ltd., Kissei Pharmaceutical Co., Ltd., Novo Nordisk Pharma Ltd., and Daiichi Sankyo Company, Ltd. K.K., T.Y., M.I., M.S., and H.K. are employees of Mitsubishi Tanabe Pharma Corporation. K.F. has no disclosures to report.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Inagaki, N., Kondo, K., Yoshinari, T. et al. Pharmacokinetic and Pharmacodynamic Profiles of Canagliflozin in Japanese Patients with Type 2 Diabetes Mellitus and Moderate Renal Impairment. Clin Drug Investig 34, 731–742 (2014). https://doi.org/10.1007/s40261-014-0226-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40261-014-0226-x