
Abstract
Background
BI 695501 is an FDA-approved biosimilar to adalimumab reference product (RP). VOLTAIRE-X was a randomized clinical trial to assess outcomes with a biosimilar monoclonal antibody in line with the FDA requirements for designation as an ‘interchangeable’ biosimilar.
Objective
The aim of this study was to assess whether multiple switches between adalimumab RP and BI 695501 lead to equivalent pharmacokinetics and a similar safety and immunogenicity profile compared with continuous adalimumab RP.
Methods
We conducted a phase III, double-blind, randomized controlled trial between July 19, 2017, and April 16, 2019. There were 49 investigational sites across Europe and North America. Of 323 screened patients with moderate-to-severe chronic plaque psoriasis, 259 were treated with adalimumab RP during the run-in period. Of these, 118 and 120 were randomized to the continuous or switching arms, respectively. Interventions consisted of a run-in period with adalimumab RP 80 mg subcutaneously (SC) on Day 1, then 40 mg SC every other week (EOW) Weeks 2–12. Patients were then randomized to receive adalimumab RP 40 mg EOW Weeks 14–48 (continuous arm) or BI 695501 40 mg Weeks 14 and 16, adalimumab RP 40 mg Weeks 18 and 20, and BI 695501 40 mg EOW Weeks 22 to 48 (switching arm); all interventions were given SC. Primary endpoints were pharmacokinetics parameters, area under the plasma concentration–time curve (AUCτ,30–32) and maximum observed drug plasma concentration (Cmax,30–32), measured after the third switch during the Week 30–32 dosing interval.
Results
238 patients (mean [standard deviation] age 44.9 [13.8]; 66.0% male) were treated in the switching (n = 118) or continuous arms (n = 120). Adjusted mean Cmax,30–32 was 7.08 and 7.00 μg/mL in the switching and continuous treatment arms, respectively; adjusted mean AUCτ,30–32 was 2025.8 and 1925.9 μg h/mL. Point estimate for mean ratio for AUCτ,30–32 was 105.2% (90.2% confidence interval [CI] 96.6–114.6), and 101.1% (90.2% CI 93.3–109.7) for Cmax,30–32. Both CIs were within a predefined bioequivalence range of 80.0–125.0%. Treatment-emergent adverse events led to discontinuation in 0.8% and 1.7% of patients in the switching and continuous treatment arms, and Psoriasis Area and Severity Index (PASI) scores were highly similar in the two arms across the entire trial period.
Conclusions
Pharmacokinetic equivalence was demonstrated, with highly similar efficacy and immunogenicity, and comparable safety observed in patients with chronic plaque psoriasis who received either adalimumab RP continuously or who switched between adalimumab RP and BI 695501.
Trial Registration
ClinicalTrials.gov: NCT03210259 (registered July 2017); Eudract.ema.europa.eu: 2016-002254-20.
Video abstract
Switching between adalimumab reference product and BI 695501 in patients with chronic plaque psoriasis (VOLTAIRE-X): a randomized controlled trial (MP4 9244 kb)
Similar content being viewed by others


Are pharmacokinetic equivalence and similar efficacy, safety, and immunogenicity maintained in patients with chronic plaque psoriasis who switched multiple times between adalimumab reference product (RP) and BI 695501? |
Switching three separate times between adalimumab RP and BI 695501 resulted in pharmacokinetic equivalence, highly similar efficacy and immunogenicity outcomes, and comparable safety compared with patients on continuous adalimumab RP. |
These findings support the interchangeability of BI 695501 with adalimumab RP. |
1 Introduction
Monoclonal antibodies targeting tumor necrosis factor-α, including infliximab and adalimumab, have greatly improved outcomes in patients with rheumatic diseases [1,2,3], psoriasis [4], and inflammatory bowel disease [5]. However, patient access can be limited due to high costs [6, 7]. Biosimilars are defined by the United States (US) Food and Drug Administration (FDA) as biologics that are highly similar to a reference product (RP), with no clinically meaningful differences in terms of safety, purity, and potency, notwithstanding minor differences in clinically inactive components [8]. The availability of biosimilars is intended to increase access to treatment for patients by decreasing cost.
BI 695501 (adalimumab-adbm; Cyltezo®, Boehringer Ingelheim International GmbH, Ingelheim am Rhein, Germany) is a biosimilar to adalimumab RP (Humira®; AbbVie Inc., North Chicago, IL, US). BI 695501 was bioequivalent to adalimumab RP in terms of pharmacokinetics (PK) in a study of healthy volunteers [9]. Subsequently, BI 695501 was shown to have equivalent efficacy and similar safety to adalimumab RP in a phase III randomized controlled trial (RCT) in patients with rheumatoid arthritis [10]. An extension study demonstrated continued similar efficacy and safety in patients receiving BI 695501 for up to 2 years [11]. Based on the extrapolation of indications, BI 695501 received approval from the FDA for use in seven indications not protected by exclusivity for which adalimumab RP is licensed, including chronic plaque psoriasis [12]. Equivalent efficacy and similar safety and immunogenicity of BI 695501 to adalimumab RP were demonstrated in patients with chronic plaque psoriasis in the VOLTAIRE-PSO study [13]. Based on the results from the VOLTAIRE-X RCT that are presented in this article, BI 695501 was approved by the FDA as being ‘interchangeable’ with adalimumab RP [14].
In the US, ‘interchangeability’ is a second regulatory (FDA) approval that allows a biosimilar product to be substituted for the RP without the intervention of the initial prescriber [15]. The first biosimilar to be approved by the FDA as being ‘interchangeable’ was insulin glargine-yfgn (SEMGLEE®) [16]; however, at the time that VOLTAIRE-X was completed, no monoclonal antibodies were approved as being ’interchangeable’ [17]. The FDA criteria for this designation require that a product must have demonstrated biosimilarity to the RP and be expected to produce the same clinical result as the RP in any given patient. Additionally, the risk in terms of safety or diminished efficacy of switching between the biosimilar and the RP should not be greater than the risk of using the RP without switching [15].
In this article, we describe the results from the VOLTAIRE-X RCT, reporting outcomes with adalimumab biosimilar in a trial designed to meet the FDA criteria for ‘interchangeability’ [15].
2 Methods
2.1 Study Design and Participants
VOLTAIRE-X (NCT03210259) was a phase III, double-blind, active comparator RCT conducted at 49 investigational sites in Europe and North America (Fig. 1). The trial design incorporated a 14-week run-in period of treatment with the adalimumab RP followed by a randomized, double-blind, 2-arm period of 34 weeks. A pre-specified blinded interim analysis with sample size reassessment was planned to be performed after approximately 86 patients with evaluable PK data reached their Week 32 endpoints assessment.

Study design. aAdalimumab RP 80 mg loading dose on Day 1 and then 40 mg/0.8 mL or 40 mg/0.4 mL EOW from Week 2 to Week 12. bFirst PK sampling interval (adalimumab RP only). cBI 695501 40 mg/0.8 mL at Week 14 and Week 16, adalimumab RP 40 mg/0.8 mL EOW at Week 18 and Week 20, and then BI 695501 40 mg/0.8 mL EOW from Week 22 to Week 48. dAdalimumab RP 40 mg/0.8 mL EOW from Week 14 to Week 48. ePrimary endpoints assessment period, and the second PK sampling interval (Week 30–32) in switching (BT) and continuous (BR) arms. BSA body surface area, EOW every other week, PASI psoriasis area and severity index, RP reference product, sPGA Static Physician’s Global Assessment
Patients were aged ≥ 18 and < 80 years at screening with a diagnosis of moderate-to-severe chronic plaque psoriasis. Patients must have had stable disease per the opinion of the investigator for 2 months prior to the first dose of study treatment, with no changes in skin morphology or significant flares at screening and baseline, as well as being considered candidates for systemic therapy or phototherapy. Full inclusion and exclusion criteria are listed in the electronic supplementary material (ESM).
The protocol was approved by the applicable independent ethics committee or institutional review board at each participating site, and the trial was performed in accordance with Good Clinical Practice and the Declaration of Helsinki. Written informed consent was obtained from all patients before their participation.
2.2 Randomization
Investigators enrolled patients, and randomization was performed during a standard pre-treatment call using an interactive response technology system. After the 14-week run-in period with adalimumab RP, patients with a ≥ 50% reduction in the Psoriasis Area and Severity Index (PASI50) were randomized 1:1 at Week 14 to either the continuous or switching treatment arms, stratified by level of response (≥ PASI50 to < PASI75, or ≥ PASI75). Patients were randomized sequentially, using the lowest available randomization number. Patients with a < 50% PASI50 response at Week 14 were discontinued from further study treatment and followed only for safety. Blinded third-party personnel administered study treatment. The interactive response technology system ensured that no person directly involved in the conduct of the trial had access to treatment allocation, except for the unblinded pharmacist and the study medication administrator. All personnel involved in trial analyses were unblinded after database lock.
2.3 Intervention
The 14-week run-in period consisted of treatment with adalimumab RP (40 mg/0.8 mL or 40 mg/0.4 mL formulation) 80 mg on Day 1, and 40 mg every other week (EOW) during Weeks 2–12, administered subcutaneously (SC). The US-licensed reference product was used across all locations (including those in Europe). Treatment in the switching arm consisted of BI 695501 40 mg (40 mg/0.8 mL formulation) at Weeks 14 and 16, followed by adalimumab RP 40 mg at Weeks 18 and 20, and then BI 695501 40 mg EOW during Weeks 22–48, all administered SC. The continuous treatment arm was adalimumab RP 40 mg EOW during Weeks 14–48. The duration of the run-in period and the number and duration of the switching intervals were aligned with the FDA, as now specified in the FDA guidance regarding study design for demonstration of interchangeability [15].
2.4 Clinical and Laboratory Monitoring
PK and immunogenicity were assessed at baseline and Weeks 2, 8, 12, 14, 18, 22, 30, 32, 40, 50, and 58. PK assessments were performed pre-dose at all visits, except for Weeks 50 (end of treatment) and 58 (end of safety follow-up period), with additional sampling 72, 120, 168, and 240 hours after Week 12 and Week 30 dosing. PASI and static Physician’s Global Assessment (sPGA) were assessed at screening, baseline, and Weeks 4, 8, 14, 18, 22, 28, 32, 40, and 50. Adverse events (AEs) were coded using the MedDRA Version 22.1. System organ classes were sorted by internationally agreed European Medicines Agency system organ class and preferred terms.
2.5 Outcomes
The primary endpoints were PK parameters area under the plasma concentration–time curve (AUCτ,30–32) and maximum observed drug plasma concentration (Cmax,30–32), measured after the third switch during the Week 30–32 dosing interval. The number and lengths of the switching intervals were in accordance with the FDA requirements [15]. Secondary PK endpoints were minimum observed plasma concentration (Cmin,30–-32) and time to maximum observed plasma concentration (tmax,30–32) assessed during Week 30–32.
Further secondary endpoints included the proportion of patients with PASI75 responses and the proportion of patients with sPGA ≤ 1 (clear or almost clear) at Week 32, in addition to the proportion of patients with antidrug antibodies (ADAs) and neutralizing antidrug antibodies (nAbs) at Week 32. Safety was assessed based on the proportion of patients with treatment-emergent adverse events (TEAEs) after randomization and during the 10-week safety follow-up period. The proportions of patients with TEAEs, serious AEs, AEs of special interest, and injection-site reactions were also assessed.
2.6 Sample Size Calculation
An initial sample size of 170 PK-evaluable patients was determined for the primary analysis at Week 32 to provide approximately 90% power, with a one-sided alpha level of 0.049, an assumed treatment difference of 8% (i.e., that the geometric mean ratio would be 92%), and a coefficient of variation of approximately 30% for Cmax,30–32 and AUCτ,30–32, respectively. These assumptions were based on data from a previous study of BI 695501 in healthy participants [9], and on simulations accounting for multiple dosing regimens and baseline PK parameters. It was assumed that approximately 30% of patients would not be evaluable for the primary analysis, due to drop-out or non-evaluable PK readout. Therefore, 240 patients were recommended to be recruited in the trial.
2.7 Statistical Analysis
Analyses of the primary and secondary PK endpoints were performed on the PK set (PKS; Suppl. Table 1, see ESM). An ANCOVA model was used to analyze AUCτ,30–32 and Cmax,30–32, which accounted for the (pre-randomization) covariables treatment (switching vs continuous arms), logarithm of PASI improvement (the ratio of PASI response at Week 14 and at Week 1), weight at Week 14, study stage (prior to or after the blinded sample size reassessment), and AUCτ,12–14 or Cmax,12–14. The blinded interim analysis was conducted and a significance level of 4.9% was used for the final analysis of the ratios of the means of AUCτ,30–32 and Cmax,30–32 across the two arms, which included a two-sided 90.2% confidence interval (CI). Equivalence was concluded if the 90.2% CI for the ratio of means across the two arms was within the bioequivalence range of 80.0–125.0%, as per FDA guidelines [15]. The secondary PK endpoint of Cmin,30–32 was analyzed in the same way as the primary endpoints; however, only the mean ratio and CI were calculated, and no formal comparison to the bioequivalence range was performed. tmax,30–32 was analyzed descriptively.
All efficacy analyses were based on the per-protocol set (Suppl. Table 1, see ESM). Confidence intervals for the differences in the proportions of patients with PASI responses in each arm were obtained using the Wald method. Safety was assessed in the safety evaluation set (treated set), while all patients treated during the run-in period were assessed in the run-in treated set (Suppl. Table 1, see ESM). Safety analyses were descriptive in nature and based on trial period. Immunogenicity assessments were descriptive in nature and were conducted in the treated set and run-in treated set.
2.8 Role of the Funding Source
Boehringer Ingelheim was involved in trial design, data collection, data analysis, and data interpretation. The authors had full access to all the data in the trial and had final responsibility for the decision to approve this manuscript for publication.
3 Results
3.1 Participants
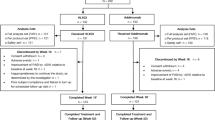
In total, 323 patients were screened, and 259 patients were treated with adalimumab RP during the run-in period (Fig. 2). Of these, 238 patients were randomized with 118 assigned to switching and 120 to continuous treatment. The per-protocol set included all 118 patients in the switching arm and 119 (99.2%) in the continuous treatment arm. The pharmacokinetic assessment set included 104 (88.1%) of the patients in the switching arm and 99 (82.5%) of those in the continuous treatment arm. Thirty-two (13.4%) patients discontinued prematurely from randomized treatment with the most common reason being withdrawal of consent. The most common reason for exclusion from the PK analysis set was missed doses, occurring in four patients (3.4%) in the switching arm and ten patients (8.3%) in the continuous arm. The first patient was enrolled on July 19, 2017, and the last patient completed treatment on April 16, 2019. Patient demographics and disease characteristics at baseline were well balanced between the two treatment arms (Table 1).

CONSORT diagram of patient flow. AE adverse events, RP reference product
3.2 Pharmacokinetics
For the primary PK endpoints, adjusted mean AUCτ,30–32 was 2025.8 and 1925.9 μg h/mL in the switching and continuous treatment arms, respectively, and adjusted mean Cmax,30–32 was 7.08 and 7.00 μg/mL, respectively (Fig. 3). Point estimates for the adjusted mean ratios of switching and continuous treatments for AUCτ,30–32 and Cmax,30–32 were 105.2% and 101.1%, respectively. The 90.2% CI ranged from 96.6 to 114.6% and 93.3 to 109.7% for the respective measures; both were within the bioequivalence range of 80.0–125.0%.

Area under the drug plasma concentration–time curve and maximum observed drug plasma concentration during the dosing interval Week 30–32 (pharmacokinetic seta)b. aAll patients who received study treatment and for whom at least one primary pharmacokinetic parameter was available. bAnalyzed using an ANCOVA model, accounting for the impact of treatment (switching vs continuous arms), logarithm of PASI improvement (the ratio of PASI response at Week 14 and at Week 1), weight at Week 14, stage (prior to or after the blinded sample size reassessment), and AUCτ,12–14 or Cmax,12–14. AUC area under the concentration–time curve, CI confidence interval, Cmax maximum observed adalimumab plasma concentration, PASI Psoriasis Area and Severity Index
Adjusted mean Cmin,30–32 was 4.91 μg/mL in the switching arm and 4.58 μg/mL in the continuous arm (point estimate for the ratios of the adjusted mean 107.3%; 90.2% CI 97.3–118.4). Median tmax,30–32 was 72.7 h (interquartile range 71.1–120.0) in the switching arm and 72.3 hours (interquartile range 70.9–120.0) in the continuous arm. Mean plasma concentration–time profiles in the two arms were highly similar over the entire study period (Suppl. Fig. 1, see ESM).
3.3 Efficacy
The proportions of patients with PASI75 responses at Week 32 were highly similar between the two arms—switching and continuous (100/118 [84.8%] and 94/119 [79.0%] patients, respectively; difference 5.8%; 90% CI − 2.5 to 14.0). PASI scores over time were highly similar in the two arms across the entire trial period (Suppl. Fig. 2, see ESM).
The proportions of patients with sPGA responses ≤ 1 at Week 32 were highly similar between the switching (83/112 patients [70.3%]) and continuous (77/109 patients [64.7%]) arms (difference 5.6%; 90% CI − 4.4 to 15.6).
3.4 Immunogenicity
Throughout the entire trial period, highly similar proportions of patients in the two treatment arms were ADA- or nAb-positive (Suppl. Fig. 3, see ESM). At Week 32, highly similar proportions of patients developed ADAs in the switching (101/112 [90.2%]; median titer 64) and continuous arms (104/110 [94.5%]; median titer 128). Results were highly similar in each arm for nAbs (switching: 46/112 [41.1%]; continuous: 46/110 [41.8%]).
3.5 Safety
During the run-in period with adalimumab RP, 103 patients (39.8%) experienced at least one TEAE, the majority of which were not serious (101 patients [39.0%]). Two patients (0.8%) experienced at least one TEAE that led to discontinuation of adalimumab RP, and one patient (0.4%) died (diffuse axonal injury and demyelination—see ESM for details). Twenty-four patients (9.3%) experienced TEAEs during this period, which were only serious in two (0.8%).
Post-randomization, 67 patients (56.8%) in the switching arm and 75 patients (62.5%) in the continuous arm experienced at least one TEAE (Table 2). The proportion of patients with a given TEAE was highly similar in the two treatment arms, except for nasopharyngitis (higher incidence in the switching arm; Suppl. Table 2, see ESM). Treatment-emergent injection-site reactions occurred in three patients (2.5%) in the continuous arm and five patients (4.2%) in the switching arm. Most TEAEs were not serious (139/238; 58.4%).
In the continuous and switching arms, 22 (18.3%) and 14 (11.9%) patients, respectively, experienced TEAEs that investigators considered related to study treatment. Across both arms, the only treatment-related TEAEs that were reported in ≥ 1% of patients were injection-site erythema (6/238; 2.3%) and arthralgia (3/238; 1.3%).
Fewer than 5% of patients experienced severe TEAEs, AEs of special interest, or TEAEs resulting in treatment discontinuation, with no notable difference between treatment groups. No patients died during the post-randomization treatment period.
4 Discussion
In VOLTAIRE-X, primary and secondary PK endpoints were equivalent and clinical outcomes were highly similar in patients with moderate-to-severe chronic plaque psoriasis who received either adalimumab RP continuously or in those who switched three times between BI 695501 and adalimumab RP. For the primary PK endpoints of Cmax,30–32 and AUCτ,30–32, the mean ratio point estimates were close to 100%, with 90.2% CIs well within the bioequivalence range of 80.0–125.0% [15, 18]. These results were supported by the secondary PK endpoints, in addition to previous studies that demonstrated highly similar PK of BI 695501 to adalimumab RP [10, 11, 19]. Although analyses of clinical efficacy were descriptive in nature, the proportions of patients with PASI75 and sPGA ≤ 1 responses at Week 32 were highly similar between the switching and continuous treatment arms. PASI responses were comparable to other studies that compared adalimumab RP with other candidate biosimilars, as well as in the VOLTAIRE-PSO study [13, 20]. Analyses of ADAs and of nAbs demonstrated comparable immunogenicity in both arms. Safety results were similar between the two arms, with no new safety signals noted. On the basis of these data, BI 695501 was approved by the FDA as being ‘interchangeable’ with adalimumab RP, becoming the first monoclonal biosimilar to be granted this designation [14]. Based on extrapolation of indications, these results provide rheumatologists, dermatologists, and gastroenterologists with data to support the substitution of BI 695501 for adalimumab RP in patients with any of the seven indications for which BI 695501 is approved [12].
While there has been speculation that multiple switches might increase immunogenicity [21], this RCT indicated highly similar immunogenicity in patients who underwent multiple treatment switches between adalimumab RP and BI 695501 compared with those who received continuous treatment with adalimumab RP. A high-sensitivity assay was used in this RCT to detect ADAs and nAbs, which resulted in positive rates of > 90% and > 41%, respectively, in both arms. Similar high levels of immunogenicity have been reported in many trials of adalimumab RP and of other candidate adalimumab biosimilars [22]. However, higher levels of immunogenicity result from the recent use of more sensitive assays than were used during the development of adalimumab RP [22, 23], but are not indicative of a change in the clinical impact of ADAs (increased drug clearance and lower drug exposure [24]), as was confirmed by the demonstration of PK bioequivalence and similar levels of immunogenicity in the two treatment arms in this RCT.
The ‘interchangeable’ designation is unique to the US regulatory system, with no equivalent in Europe and other regions. An FDA-approved interchangeable biosimilar can be substituted for its reference product by a pharmacist without requiring prior approval from the prescriber (where permitted by state law) [15]. This designation of interchangeability is only applicable to the individual biosimilar approved by the FDA as interchangeable; pharmacist-level substitution can occur between the interchangeable biosimilar and the RP, but not between the RP and its other biosimilars, or between the interchangeable biosimilar and other biosimilars of the RP.
This RCT of a biosimilar monoclonal antibody satisfies the new FDA requirements for demonstrating interchangeability with an RP, although the guidance for interchangeability had not yet been published at the time the protocol was being developed [15]. This RCT assessed PK parameters as primary endpoints, and immunogenicity, efficacy, and safety were secondary endpoints, consistent with FDA guidance for demonstrating interchangeability [15]. Accordingly, three switches between adalimumab RP and BI 695501 were included in the switching arm. This RCT was also conducted in an indication considered sensitive for the assessment of PK bioequivalence, as adalimumab is administered as monotherapy in patients with plaque psoriasis [25]. In contrast, patients with rheumatoid arthritis frequently receive concomitant treatment with methotrexate, which could potentially reduce the immunogenicity of adalimumab and alter its PK.
A final strength of this RCT was that it included a ‘run-in’ period to account for inter-patient variability of adalimumab exposure [26] and to generate an additional covariate for the ANCOVA model used for the analysis of the primary PK endpoints.
Three switches were mandated in the switching arm, as is now specified in the FDA guidance document [15]. In clinical practice, patients may be subjected to more than three switches between the RP and a biosimilar, be treated with one agent for a longer duration before switching, be switched between different available biosimilars, or from a biosimilar back to the RP. However, it is not possible to assess all of the potential scenarios that might occur in clinical practice in a single RCT. A further minor limitation of this RCT was the reduced precision of tmax and Cmax assessments as a consequence of sparse PK sampling, due to clinical feasibility of sampling in an outpatient setting. Nonetheless, a possible small bias in Cmax was not expected to affect the assessment of the primary PK endpoints and the conclusion of bioequivalence, as both arms were affected in the same way.
5 Conclusion
VOLTAIRE-X met FDA requirements for demonstrating interchangeability of a biosimilar monoclonal antibody with its RP. Based on equivalent PK and highly similar efficacy, immunogenicity, and safety in the two treatment arms, BI 695501 has been approved by the FDA as being ‘interchangeable’ with adalimumab RP, becoming the first monoclonal biosimilar to be granted this designation [14].
Availability of data and materials
To ensure independent interpretation of clinical study results, Boehringer Ingelheim grants all external authors access to relevant material, including participant-level clinical study data, as needed by them to fulfill their role and obligations as authors under the ICMJE criteria. Clinical study documents and participant clinical study data are available to be shared on request after publication of the primary manuscript in a peer-reviewed journal, and if regulatory activities are complete and other criteria met as per the BI Policy on Transparency and Publication of Clinical Study Data (see https://www.mystudywindow.com/us/). Bona fide, qualified scientific and medical researchers are eligible to request access to the clinical study data with corresponding documentation describing the structure and content of the datasets. Upon approval, and governed by a Legal Agreement, data are shared in a secured data-access system for a limited period of 1 year, which may be extended upon request. Prior to providing access, clinical study documents and data will be examined, and, if necessary, redacted and de-identified, to protect the personal data of study participants and personnel, and to respect the boundaries of the informed consent of the study participants. Researchers should use the https://vivli.org/ link to request access to study data and visit https://www.mystudywindow.com/us/ for further information.
Code availability
Not applicable.
Change history
31 October 2022
A peer-reviewed video abstract was retrospectively added to this publication.
References
Dörner T, Strand V, Castaneda-Hernandez G, et al. The role of biosimilars in the treatment of rheumatic diseases. Ann Rheum Dis. 2013;72(3):322–8.
Dörner T, Strand V, Cornes P, et al. The changing landscape of biosimilars in rheumatology. Ann Rheum Dis. 2016;75(6):974–82.
Strand V, Kimberly R, Isaacs JD. Biologic therapies in rheumatology: lessons learned, future directions. Nat Rev Drug Discov. 2007;6(1):75–92.
Ellis AG, Flohr C, Drucker AM. Network meta-analyses of systemic treatments for psoriasis: a critical appraisal. Br J Dermatol. 2019;180(2):282–8.
Rawla P, Sunkara T, Raj JP. Role of biologics and biosimilars in inflammatory bowel disease: current trends and future perspectives. J Inflamm Res. 2018;11:215–26.
Dranitsaris G, Jacobs I, Kirchhoff C, Popovian R, Shane LG. Drug tendering: drug supply and shortage implications for the uptake of biosimilars. Clinicoecon Outcomes Res. 2017;9:573–84.
Smeeding J, Malone DC, Ramchandani M, Stolshek B, Green L, Schneider P. Biosimilars: considerations for payers. Pharm Ther. 2019;44(2):54–63.
FDA. Biosimilar and Interchangeable Products. 2017. https://www.fda.gov/drugs/biosimilars/biosimilar-and-interchangeable-products. Accessed May 2021.
Wynne C, Altendorfer M, Sonderegger I, et al. Bioequivalence, safety and immunogenicity of BI 695501, an adalimumab biosimilar candidate, compared with the reference biologic in a randomized, double-blind, active comparator phase I clinical study (VOLTAIRE®-PK) in healthy subjects. Expert Opin Investig Drugs. 2016;25(12):1361–70.
Cohen SB, Alonso-Ruiz A, Klimiuk PA, et al. Similar efficacy, safety and immunogenicity of adalimumab biosimilar BI 695501 and Humira reference product in patients with moderately to severely active rheumatoid arthritis: results from the phase III randomised VOLTAIRE-RA equivalence study. Ann Rheum Dis. 2018;77(6):914–21.
Cohen SB, Czeloth N, Lee E, Klimiuk PA, Peter N, Jayadeva G. Long-term safety, efficacy, and immunogenicity of adalimumab biosimilar BI 695501 and adalimumab reference product in patients with moderately-to-severely active rheumatoid arthritis: results from a phase 3b extension study (VOLTAIRE-RAext). Expert Opin Biol Ther. 2019;19(10):1097–105.
FDA. Cyltezo® Prescribing Information. 2017. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761058lbl.pdf. Accessed 5 Oct 2021.
Menter A, Arenberger P, Balser S, et al. Similar efficacy, safety, and immunogenicity of the biosimilar BI 695501 and adalimumab reference product in patients with moderate-to-severe chronic plaque psoriasis: results from the randomized Phase III VOLTAIRE-PSO study. Expert Opin Biol Ther. 2021;21(1):87–96.
FDA. Cyltezo® press release. 2021. https://www.fda.gov/news-events/press-announcements/fda-approves-cyltezo-first-interchangeable-biosimilar-humira. Accessed Oct 2021.
FDA. Considerations in demonstrating interchangeability with a reference product. 2019. https://www.fda.gov/media/124907/download. Accessed June 2021.
FDA. SEMGLEE® Prescribing Information. 2021. https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761201Orig1s000lbl.pdf. Accessed Oct 2021.
Center for Drug Evaluation and Research. List of licensed biological products with (1) reference product exclusivity and (2) biosimilarity or interchangeability evaluations to date. https://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/UCM560162.pdf. Accessed June 2021.
Alvarez DF, Wolbink G, Cronenberger C, Orazem J, Kay J. Interchangeability of biosimilars: what level of clinical evidence is needed to support the interchangeability designation in the United States? BioDrugs. 2020;34:723–32.
Kang J, Eudy-Byrne RJ, Mondick J, Knebel W, Jayadeva G, Liesenfeld KH. Population pharmacokinetics of adalimumab biosimilar adalimumab-adbm and reference product in healthy subjects and patients with rheumatoid arthritis to assess pharmacokinetic similarity. Br J Clin Pharmacol. 2020;86(11):2274–85.
Wan MT, Alvarez J, Shin DB, Dommasch ED, Wu JJ, Gelfand JM. Head-to-head trials of systemic psoriasis therapies: a systematic review of study design and maximum acceptable treatment differences. J Eur Acad Dermatol Venereol. 2019;33(1):42–55.
Numan S, Faccin F. Non-medical switching from originator tumor necrosis factor inhibitors to their biosimilars: systematic review of randomized controlled trials and real-world studies. Adv Ther. 2018;35(9):1295–332.
Strand V, Gonçalves J, Hickling TP, Jones HE, Marshall L, Isaacs JD. Immunogenicity of biosimilars for rheumatic diseases, plaque psoriasis, and inflammatory bowel disease: a review from clinical trials and regulatory documents. BioDrugs. 2020;34(1):27–37.
Strand V, Goncalves J, Isaacs JD. Immunogenicity of biologic agents in rheumatology. Nat Rev Rheumatol. 2021;17(2):81–97.
Mostafa NM, Nader AM, Noertersheuser P, Okun M, Awni WM. Impact of immunogenicity on pharmacokinetics, efficacy and safety of adalimumab in adult patients with moderate to severe chronic plaque psoriasis. J Eur Acad Dermatol Venereol. 2017;31(3):490–7.
Alwawi EA, Mehlis SL, Gordon KB. Treating psoriasis with adalimumab. Ther Clin Risk Manag. 2008;4(2):345–51.
Ward MG, Thwaites PA, Beswick L, et al. Intra-patient variability in adalimumab drug levels within and between cycles in Crohn’s disease. Aliment Pharmacol Ther. 2017;45(8):1135–45.
Acknowledgements
The authors meet criteria for authorship as recommended by the International Committee of Medical Journal Editors (ICMJE). The authors received no direct compensation related to the development of the manuscript. Writing and editorial support for this manuscript were provided by Craig Turner, of Ashfield MedComms, an Ashfield Health company, which was contracted and funded by Boehringer Ingelheim Pharmaceuticals Inc. (BIPI), for these services. BIPI was given the opportunity to review the manuscript for medical and scientific accuracy as well as intellectual property considerations.
Funding
This trial was funded by Boehringer Ingelheim International GmbH, Ingelheim, Germany.
Author information
Authors and Affiliations
Contributions
Conception and design: K.H.L., B.L., S.B. Analysis and interpretation of the data: all authors. Drafting of the article: all authors. Critical revision of the article for important intellectual content: all authors. Final approval of the article: all authors. Collection and assembly of data: B.L.
Corresponding author
Ethics declarations
Conflict of interest
Alan Menter received grant support from AbbVie, Argenx, Novartis, Pfizer, Sun Pharma, and UCB; he reported consulting fees from AbbVie, Amgen, Boehringer Ingelheim, Biocon, Janssen Biotech, LEO Pharma, Novartis, Sun Pharma, and UCB; honoraria for lectures/presentations/speaker bureaus from AbbVie, Amgen, Eli Lilly, Janssen, Sun Pharma, and UCB; travel support from Boehringer Ingelheim, Eli Lilly, and Sun Pharma; and participated in data safety monitoring/advisory boards for Amgen and Boehringer Ingelheim. Stanley Cohen received grant support from AbbVie, Amgen, BMS, Genentech, Lilly, Pfizer, and Roche; he reported consulting fees from AbbVie, Aclaris, Amgen, Boehringer Ingelheim, Genentech, and Pfizer; honoraria from Novartis and Pfizer; and participated in a data safety monitoring board for Gilead Sciences. Jonathan Kay received grant support from Aker BioMarine, Alliance for Lupus Research, AMPEL BioSolutions, Gilead Sciences, Novartis, Pfizer, and UCB; he reported royalty fees from UpToDate; consulting fees from AbbVie, Alvotech, Boehringer Ingelheim, Celltrion Healthcare, Horizon Therapeutics, Jubilant Radiopharma, Merck Sharp & Dohme, Mylan, Novartis, Pfizer, Samsung Bioepis, Sandoz, Scipher Medicine, and UCB; and participated in data safety/advisory boards for Bristol Myers Squibb, Inmagene, and Kolon TissueGene. Vibeke Strand reported consulting fees from AbbVie, Amgen, Arena, Aria, AstraZeneca, Bayer, Bioventus, BMS, Boehringer Ingelheim, Celltrion, Chemocentryx, EMD Serono, Flexion, Galapagos, Genentech/Roche, Gilead, GSK, Horizon, Ichnos, Inmedix, Janssen, Kiniksa, Eli Lilly, Merck, MiMedx, Novartis, Pfizer, Regeneron, Rheos, R-Pharma, Samsung, Sandoz, Sanofi, Scipher, Servier, Setpoint, Sun Pharma, Tonix, and UCB. Alice Gottlieb received honoraria as an advisory board member and consultant for AnaptsysBio, Avotres Therapeutics, Boehringer Ingelheim, Bristol Myers Squibb Co., GSK, Janssen, Eli Lilly, Leo, Novartis, Pfizer, Sun Pharmaceuticals, UCB, Dermavant, and Xbiotech; and received research/educational grants from Boehringer Ingelheim, Janssen, Novartis, UCB, and Sun Pharmaceutical Industries, Inc. Stephen Hanauer received grant support from AbbVie, Allergan, Amgen, Celgene, Genentech, GlaxoSmithKline, Janssen, Eli Lilly, Novartis, Pfizer, Prometheus, Receptos, Takeda, and UCB; he reported consultancy fees from AbbVie, Allergan, Amgen, Arena, AstraZeneca, Boehringer Ingelheim, Bristol Myers Squibb, Celgene, Cosmos, Catalys Pacific, Covance, Genentech, GlaxoSmithKline, Janssen, Eli Lilly, Merck, Novartis, Pfizer, Progenity, Prometheus, Receptos, Salix, Samsung Bioepis, Seres Therapeutics, Sorriso, Takeda, TLL Pharma, UCB, and VHSquared; speaker fees from AbbVie, Bristol Myers Squibb, Janssen, Pfizer, and Takeda; and participated in data safety monitoring boards for Arena, Boehringer Ingelheim; Bristol Myers Squibb, Gossamer, Prometheus, and Protagonist. Susanne Buschke, Karl-Heinz Liesenfeld, Jennifer Schaible, Dorothy McCabe, and Benjamin Lang reported being employees of Boehringer Ingelheim. Sravan Kumar Eduru was previously employed by Boehringer Ingelheim.
Consent to participate
The protocol was approved by the applicable independent ethics committee or institutional review board at each participating site, and the trial was performed in accordance with Good Clinical Practice and the Declaration of Helsinki. Written informed consent was obtained from all patients before their participation.
Consent for publication
All authors have reviewed the final version of this manuscript and approved its publication.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Menter, A., Cohen, S., Kay, J. et al. Switching Between Adalimumab Reference Product and BI 695501 in Patients with Chronic Plaque Psoriasis (VOLTAIRE-X): A Randomized Controlled Trial. Am J Clin Dermatol 23, 719–728 (2022). https://doi.org/10.1007/s40257-022-00708-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40257-022-00708-w