
Abstract
Dendritic cells are central orchestrators of the immune response. They communicate with both innate and adaptive immune systems and can aggravate or attenuate renal inflammation depending on the microenvironment and the stage of disease. This review summarizes current knowledge regarding the role of dendritic cells in renal inflammation and highlights the recent advances in understanding their function in the development and resolution of various experimental and human kidney diseases. We anticipate that recent advances in this research area will yield in the near future novel therapeutic strategies targeting DCs to control acute and chronic renal injury.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Dendritic cells (DCs) were first discovered by immunologist Ralph M. Steinman in 1973, who in 2011 received a Nobel Prize in Medicine for this discovery. In recent decades, DCs have attracted great interest from researchers due to their central role in both innate and adaptive immunity.
The progression of kidney diseases characteristically involves inflammation. Innate immunity plays an important role in antigen-independent types of inflammation. DCs affect innate immunity through secretion of various cytokines and chemokines, which act on innate immune cells and renal parenchymal cells [1]. These cytokines and chemokines can be pro-inflammatory or anti-inflammatory and some have a direct pro-fibrotic effect. DCs also act as effective antigen presenting cells (APCs) to modulate adaptive immunity. Adaptive immunity plays an important role in kidney diseases that are caused by foreign or auto-antigens, such as in experimental nephrotoxic nephritis (NTN), T cell-mediated glomerulonephritis and lupus nephritis. DCs accumulate in inflamed kidneys and they can either aggravate or attenuate kidney injury. Currently, it is unclear why DCs are predominantly pro-inflammatory in some kidney diseases but anti-inflammatory in others. In this review, we will summarize current knowledge regarding the role of DCs in renal inflammation, and will highlight recent advances in understanding their function in the development and resolution of various experimental and human kidney diseases. This review will not cover the role of DCs in transplantation, which has been extensively reviewed elsewhere [2].
DC Biology & Kidney Disease
DC Origin and Subsets
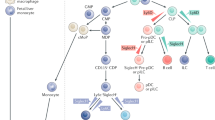
Macrophages, monocytes and DCs share a common progenitor called macrophage-DC progenitor (MDP), which is derived from the common myeloid progenitor (CMP) [3]. MDPs give rise to the common DC progenitor (CDP) which then differentiates into pre-DCs and plasmacytoid DCs [3]. Pre-DCs migrate to lymphoid tissue to differentiate into DCs. In addition, circulating monocytes derived from MDPs can also give rise to DCs.
Several different classification systems have been proposed to define DC subsets. However, the phenotypic and functional plasticity of DCs have made this difficult. DCs can be broadly classified into conventional DCs that comprise DC subsets derived from pre-DCs, and non-conventional DCs which include monocyte-derived DCs along with plasmacytoid DCs [3]. In murine kidneys, the surface marker CD11c and high expression of MHC class II were previously used to identify DCs and to distinguish them from macrophages. However, recent studies showed that cells of the renal mononuclear phagocytic system can simultaneously express cell surface markers traditionally associated with either DCs or macrophages, indicating that there is substantial functional and phenotypic overlap between these cells [4, 5]. In humans, CD11c is also expressed by monocytes and macrophages [6]. Markers such as CD1c/BDCA-1, CD141/BDCA-3 and CD209/DC-SIGN have been used to identify DC subtypes in human kidneys.
DC Maturation and Cytokine Profile
In normal kidneys, DCs form a contiguous assembly throughout the interstitium, acting as an anatomical surveillance network [7]. They are present in their immature form, expressing low levels of MHC class II and co-stimulatory molecules [8]. These immature DCs have high phagocytic activity, allowing them to constantly sample antigens and to present these antigens to T cells in draining lymph nodes [8]. In the absence of maturation stimuli such as damage-associated molecular patterns (DAMPs), microbe-associated molecular patterns (MAMPs), various cytokines and chemokines, these DCs tolerize autoreactive T cells to help maintain peripheral tolerance [9]. In contrast, in the presence of the aforementioned stimuli, DCs undergo functional maturation. These matured DCs exhibit limited phagocytic activity, express high levels of MHC class II and co-stimulatory molecules, secrete large quantities of cytokines and chemokines and induce both humoral and cellular adaptive immune responses [8].
DCs can secrete a variety of cytokines, some of which are pro-inflammatory whilst others are anti-inflammatory. For example, DCs are the predominant secretor of pro-inflammatory cytokine TNF-α in the first 24 h post ischemia reperfusion injury (IRI) [10]. DCs also secrete IL-12 in later stage NTN and lupus nephritis which promotes inflammation [11••, 12]. On the other hand, DCs secrete the anti-inflammatory cytokine IL-10 in cisplatin nephrotoxicity (CNT) and early stage NTN [13, 14].
Chemokine Receptors and DC Migration
There are four families of chemokines, C, CC, CXC and CX3C. Eighteen chemokine receptors have been identified to date [15]. Amongst these, particular chemokines and their receptors guide the migration of DCs into tissue. For example, CCR7, CXCR4, CCR2 and CXCR5 on DCs play a role in their migration to the draining LNs [15]. In recent years, targeting these chemoreceptors to inhibit migration of immune cells has attracted great attention.
CCR1, CCR2, CCR5, CX3CR1 and CCR7 on DCs have been shown to contribute to their entry into healthy kidney [16]. Chemokine receptor CX3CR1 expressed by DCs has been identified recently by Hochheiser et al. as a specific receptor that can be used as a therapeutic target for treating progressive kidney disease. This chemokine receptor is expressed by DCs and monocytes. It has only one ligand CX3CL1 which is presented by various tissues. The authors found that in kidneys, this chemokine receptor targeted kidney DCs with relative specificity [17••]. In steady state, the abundance of DCs, but not macrophages was dependent on CX3CR1. Of the organs tested, kidneys were shown to express the highest levels of CX3CL1. The authors then used the NTN model to study the effect of CX3CR1 on kidney DCs under inflammatory conditions. They found that CX3CR1 mediated DC recruitment into kidneys. CX3CR1 deficiency significantly reduced DC numbers, especially in the kidney cortex and attenuated NTN [17••]. Therefore, targeting CX3CR1 may represent a more selective therapeutic approach in treating kidney diseases, with fewer side effects compared with traditional immunotherapy which is usually non-specific. However, more studies are required to assess the benefit and side effects of targeting this chemokine receptor to treat kidney disease.
Innate Immunity, DAMPs and Dendritic Cells
Kidney diseases in which innate immunity plays a predominant role involve sterile inflammation. Sterile inflammation is triggered by DAMPs which are derived from either intracellular sources such as nuclear proteins (e.g. high-mobility group box 1 [HMGB-1]), mitochondrial components, uric acid, cellular chaperones (e.g. heat shock proteins), or extracellular matrix moieties including hyaluronan [18]. DAMPs are recognized by pattern-recognition receptors (PRRs) such as Toll-like receptors (TLRs) and NOD-like receptors (including the NLRP inflammasomes) [19]. DCs play an important role in this process. For example, TLR2 and TLR4 on DCs have been implicated in IRI, and the NLRP3 cascade in DCs has been implicated in crystal nephropathy. Details of these processes will be discussed in subsequent sections.
The Role of DCs and T Cells in Glomerulonephritis
Glomerulonephritis has been traditionally viewed as a disease caused by immune complex deposition followed by complement activation. Increasing evidence over recent years has suggested that T cells and DCs play an important role in this disease. T cells have long been known to help B cells to produce antibodies in glomerulonephritis. Recent studies have shown that they also act as immune effector cells. For example, in NTN, DCs presented filtered antigen to T helper (Th) cells and stimulated them to produce pro-inflammatory cytokines such as IL-2, IL-6, IL-17, IFN-γ and TNF-α, and exacerbated NTN [11••]. DCs recruited and activated Th1 cells and these activated Th1 cells recruited more pro-inflammatory cells to form periglomerular infiltrates [20]. In the nephrin-OVA-HEL (NOH) mouse model developed to study T cell-mediated glomerulonephritis, DCs captured glomerular antigens and presented them to Th cells leading to cytokine and chemokine production by both cells [21]. This resulted in the recruitment of more immune effector cells including T cells and macrophages, which formed periglomerular infiltrates resembling the infiltrates present in NTN [21, 22]. In both of these models, depletion of DCs reduced interstitial infiltrates and attenuated disease. DCs are shown to be located mainly in the tubulointerstitium [23]. These findings demonstrated that cross talk between T cells and DCs contributed to the spreading of glomerular injury to the tubulointerstitium.
DCs in Experimental Models of Kidney Disease
DCs in Ischemia Reperfusion Injury
Renal ischemia reperfusion injury is an important cause of acute kidney injury (AKI). Inflammation plays a key role in the pathophysiology of IRI [24, 25]. Ischemia and/or reperfusion results in loss of immune system homeostasis leading to inflammation and AKI [25]. Innate immunity is the predominant immune response, but adaptive immunity also plays a role. In IRI, the acute reduction of blood flow resulted in hypoxia-induced vascular and tubular dysfunction [26]. Vascular endothelial cells were activated by hypoxia which led to enhanced DC adhesion and migration [27]. It has been shown that following IRI, DCs were amongst the earliest producers of pro-inflammatory cytokines and chemokines such as IL-6, TNF-α, RANTES and MCP-6 [10]. In the first 24 h post IRI, DCs were the predominant secretors of TNF-α [10], which is both pro-inflammatory and pro-fibrotic [28–30].
The mechanism by which innate immunity is triggered by IRI has been studied. During IRI, damaged tissues released DAMPs such as histone, heat shock proteins, HMGB-1 and extracellular matrix components such as hyaluronan, fibronectin and biglycan, which bound to and activated TLR2 and TLR4 on renal DCs [31–35]. These activated DCs then secreted pro-inflammatory cytokines and chemokines which triggered leukocyte recruitment resulting in renal inflammation [31].
DCs promote innate immune response also through interaction with natural killer T (NKT) cells following IRI. Studies have shown that CD1d molecules expressed on kidney DCs can present endogenous glycolipid to NKT cells, inducing NKT cell activation to promote inflammation [26]. Tolerizing DCs with an ex vivo adenosine 2A receptor agonist suppressed NKT cell activation in vivo and attenuated AKI [36•].
In addition, DCs also have a role to play in adaptive immune response post IRI. DCs responded to IRI by increasing trafficking of protein antigens from kidney to renal lymph nodes (RLNs), resulting in an increased activation of localized antigen-specific CD4 + T cells [37].
In contrast to the above pro-inflammatory role, the anti-inflammatory role of DCs in IRI has also been demonstrated in multiple studies. Ischemia activated innate immune system through TLR signalling such as that of TLR4 on the DCs. It was shown that single Ig/IL-1–related receptor (SIGIRR), which was constitutively expressed in intrarenal DCs, inhibited TLR signalling in DCs, suppressed its activation and thereby limited sterile inflammation post IR [38]. SIGIRR-deficient kidneys had increased leucocyte infiltrates and worse post-ischemic AKI compared with wild-type IR kidneys [38]. Thus, SIGIRR inhibited inappropriate immune activation post IR to prevent unnecessary tissue damage by suppressing activation of DCs [38]. The same group subsequently showed that IR can also induce inhibitors such as IFN Regulatory Factor 4 in DCs which suppressed the secretion of TNF-α [39].
In ischemic preconditioning, which has an anti-inflammatory effect and provides protection against renal IRI, DCs were shown to be one of the major mediators, and depletion of DCs was associated with partial loss of preconditioning benefits [40].
DCs also exert their protective effect in IRI through interaction with mesenchymal stem cells (MSC). MSC are multi-potent progenitor cells which have been shown to have renoprotective effects post IRI [41]. This renoprotective effect was partially mediated through interaction with DCs, which was in part dependent on IL-10 [42•].
During the recovery phase of IRI, DCs changed from a pro-inflammatory to an anti-inflammatory phenotype. Kim et al. showed that depletion of DCs with clodronate post IRI was associated with an increased inflammatory milieu with more neutrophils and pro-inflammatory cytokines in the kidney and lower levels of IL-10 [43•]. Kidney CD11c + cells isolated on day 7 exhibited an anti-inflammatory phenotype with lower levels of pro-inflammatory cytokines (IL-6, TNF-α and IFN-γ) and increased production of IL-10 in comparison with those isolated on day 1 [43•].
DCs in Unilateral Ureteral Obstruction
Unilateral ureteral obstruction (UUO) is a commonly used model to study renal inflammation leading to fibrosis. Earlier studies demonstrated that UUO was associated with rapid accumulation of F4/80 + DCs which were a predominant producer of pro-inflammatory cytokines such as TNF-α during early stage UUO; F4/80 + DCs also induced IL-17 and IFN-γ expression in CD4 + T cells [44]. However, the authors used clodronate to deplete cells, which may deplete all F4/80 + cells including macrophages, and not just DCs [45]. When they used diphtheria toxin (DT) to more specifically deplete DCs in CD11c-DTR mice, it became clear that monocyte lineage cells other than DCs promoted fibrosis in UUO [46]. Another study showed that early in UUO, pro-inflammatory functions of DCs were related to their role as APCs to better stimulate T cell responses, yet they did not directly contribute to fibrosis [47].
DCs in Cisplatin Nephrotoxicity
DCs have been shown to attenuate CNT, by a mechanism which is partially mediated through production of IL-10. It was found that DCs not only produced IL-10 themselves but also expressed Inducible T cell Co-Stimulator Ligand (ICOS-L) that can interact with Tregs to secrete IL-10 [13]. Endogenous IL-10 reduced inflammation and was protective against CNT [48]. The authors then used diphtheria toxin to deplete DCs in the CNT model and found that depletion of DCs aggravated cisplatin-induced AKI [49].
DCs in crystal nephropathy
Crystal nephropathy causes AKI not only though urinary outflow obstruction, but also through substantial interstitial inflammation. It was recently found that calcium oxalate crystals activated NLRP3 inflammasome cascade in DCs, stimulating them to secrete the pro-inflammatory cytokine IL-1β, resulting in inflammation and kidney injury in crystal nephropathy [50•]. Depletion of DCs reduced intrarenal IL-1β production and attenuated crystal nephropathy [50•].
DCs in Nephrotoxic Nephritis
Nephrotoxic nephritis (NTN) is an experimental model of human crescentic glomerulonephritis, induced using sheep anti-glomerular basement membrane antiserum. The sheep antibody is taken up by APCs which then stimulate Th1 and Th17 cells to promote inflammation, resulting in a delayed type hypersensitivity kidney injury [51]. In this disease model, CD11c + DCs accumulate only in the tubulointerstitium, not in the inflamed glomeruli [52]. In vivo depletion of DCs has been performed to investigate their functional role in NTN. Scholz et al. have shown that depletion of CD11c + DCs 4 and 10 days after the induction of NTN aggravated the disease with more severe tubulointerstitial and glomerular damage, proteinuria and decrease in creatinine clearance [14]. This indicates a protective role of DCs in this disease model, which appears to be mediated by several mechanisms. Firstly, there was increased production of IL-10 which had been shown to attenuate NTN [53]. DCs not only produced IL-10 themselves, but also expressed ICOS-L which stimulated CD4 + T cells to secrete IL-10 [14]. Secondly, DCs recruited anti-inflammatory leukocytes such as invariant natural killer T (iNKT) cells to suppress harmful immune response in the early phase of NTN. iNKT cells were known to impede the development of experimental crescentic glomerulonephritis [54]. DCs recruited protective iNKT through production of cognate ligand CXCL16 early after induction of nephritis [55] and thereby attenuated glomerular inflammation.
However, DCs acquired a pro-inflammatory role at later stages of NTN. Late depletion of DCs after 7 days attenuated NTN as evidenced by a reduction in kidney infiltration by macrophages and T cells, as well as the number of crescents [11••]. It was shown that at a later stage of NTN, DCs captured more antigen (signal 1) in proteinuric mice, increased their expression of co-stimulatory molecules including CD80 and CD86 (signal 2) and increased secretion of pro-inflammatory cytokines such as IL-12 and TNF-α (signal 3) [11••]. At the same time, there was a decreased production of ICOS-L. The intensification of these three signals enhanced the ability of DCs to drive an inflammatory response. These findings suggest a switch of DCs from an early protective to a later pro-inflammatory phenotype during the course of NTN [11••].
DCs in T Cell-Mediated Glomerulonephritis
Glomerulonephritis has been characterized by immune complex deposition and complement activation [56]. It has been suggested in recent years that DCs and T cells play an important role in this disease in the absence of immune complex-mediated pathology. Heymann used a transgenic NOH mouse model to study the role of these cells in glomerulonephritis [21]. He found that co-injection of CD4 + Th and CD8 + cytotoxic T lymphocytes (CTL) which were specific for glomerular antigens can lead to formation of periglomerular mononuclear infiltrates [21]. Kidney damage was found after 4 weeks of repetitive injection [21]. DCs played a central role in this process in that they took up auto-antigens and trafficked them to RLNs where they activated CTLs [21]. Activated CTLs then entered the kidney and released more glomerular antigen for cross-presentation to CTLs in the RLNs [21]. At the same time, DCs in the tubulointerstitium also presented these auto-antigens to CD4 + T cells, leading to the production of pro-inflammatory cytokines and chemokines, and recruitment of more CTLs, DCs, and macrophages to form periglomerular infiltrates [21]. Depletion of DCs resulted in rapid dissolution of these infiltrates and attenuation of disease [21]. DCs were known to be located mainly in the tubulointerstitium [23]. These findings suggest that DCs may represent a potential link between glomerular injury and disease progression into the tubulointerstitium, and that targeting pro-inflammatory DCs may be a promising approach to delay kidney disease progression.
DCs in Experimental Lupus Nephritis
Studies using murine lupus nephritis models showed that DCs affected this disease both systemically through induction of auto-antibodies and locally by contributing to renal inflammation [57]. The systemic role of DCs in experimental lupus nephritis has been reviewed elsewhere [57] and is beyond the scope of this review. In the kidney, DCs produced IL-12 and induced a pro-inflammatory microenvironment in this disease [12]. Iwata et al. showed that DCs promoted lupus nephritis through secretion of HMGB-1, a pro-inflammatory cytokine, via activation of p38 mitogen-activated protein kinase (MAPK) [58]. DCs also contributed to local synthesis of C1q, which is a subunit of the C1 complex of the classical complement pathway [59•]. Therefore, DCs promote inflammation in lupus nephritis through secretion of pro-inflammatory cytokines and activation of complement.
DCs in Human Kidney Disease
In human kidney disease, the role of DCs has remained poorly defined due to lack of specific markers. In addition, the differences between human and mouse DC subset markers have made it difficult to compare human and mouse DCs. Nevertheless, in recent years, many DC-specific markers have been found, which may be used to identify DCs in human kidneys. The majority of these studies found that during injury, DCs accumulated in the tubulointerstitial compartment, but rarely in the glomeruli. The first study using more recent markers of DCs in human kidneys was published by Woltman et al. They analysed frozen tissues from pre-transplant biopsies, renal allograft rejection and IgA nephropathy [60]. It was found that in normal kidneys, DCs were located mainly in the tubulointerstitium. They were also seen frequently around, but rarely within glomeruli. In the biopsies from patients with acute allograft rejection and IgA nephropathy, the number of these DCs was found to be increased, suggesting an accumulation of DCs in the context of inflammation [60]. In another study, Segerer et al. found that in proliferative glomerulonephritis, DCs were seen only in the tubulointerstitium [61] with their number correlating with serum creatinine. Fiore et al. examined the biopsies of 21 patients with lupus nephritis. They found three subsets of DCs (BDCA1 + and BDCA3 + myeloid DCs, and BDCA2 + plasmacytoid DCs) with decreased circulating levels, which were, however, recruited into the kidney tubulointerstitium [62] suggesting their pathogenic role in lupus nephritis. More recently, Kassianos et al. found that there was a markedly elevated number of tubulointerstitial myeloid DCs in biopsies of kidneys with interstitial fibrosis in comparison to non-fibrotic kidneys, with myeloid DCs identified as a key producer of pro-fibrotic cytokine TGF-β [63••]. These results suggest that local and recruited DCs accumulate in tubulointerstitium in response to injury and acquire a pro-inflammatory and pro-fibrotic function to promote inflammation and interstitial fibrosis.
There are two studies showing DCs accumulating in glomeruli in human kidney disease. One found glomerular accumulation of plasmacytoid DCs in lupus nephritis [64]. The other study found immature DCs in glomeruli in renal biopsies from patients with ANCA-associated vasculitis [65]. However, this is not supported by two other studies mentioned above [61, 62]. One possible explanation for this discrepancy could be the influx of DCs from interstitium into glomeruli resulting from destruction of Bowman’s capsule which occurs quite commonly in ANCA-associated vasculitis. More studies are needed to confirm the presence of these DCs in glomeruli and to clarify their role in kidney injury.
Challenges and Future Directions
In this review, we have summarized the current progress of DC studies in experimental and human renal inflammation (Table 1, Fig. 1). DCs are a central regulator of immune response. They are positioned to form an anatomical surveillance network within the renal parenchyma. DCs constantly sample the surrounding environment and communicate with both innate and adaptive immunity. They can aggravate or attenuate renal inflammation in different kidney disease models. DCs, as central orchestrators of immune response, have attracted great interest as a therapeutic target to treat human diseases. It is also possible that they may, in the future, be used as therapeutic tools for kidney disease [66, 67].

Pro- and anti-inflammatory functions of DCs in experimental kidney disease models. CXCL16 chemokine (C-X-C motif) ligand 16; DC dendritic cell; HMGB-1 high-mobility group box 1 protein; ICOS-L inducible T cell co-stimulator ligand; IFN RF interferon regulatory factor; IL interleukin; MSC mesenchymal stem cells; iNKT invariant natural killer T cells; SIGIRR single Ig/IL-1–related receptor; TNF-α tumour necrosis factor alpha
Despite the abundant evidence of DCs accumulating in inflamed kidney tissue and their central role in renal inflammation, research in this area has been hampered by several challenges that have impeded the development of therapeutic strategies targeting DCs to treat renal inflammation. The first of these challenges is that the methods used to deplete DCs are not specific for DCs. For example, the methods to deplete DCs such as liposomal clodronate and using CD11c DTR mice both deplete non DCs including macrophages. Therefore, effects attributed to DCs that have been extrapolated from studies using these techniques could be due to functions of cells other than DCs. Secondly, there is substantial overlap across DC and macrophage populations. Traditionally, macrophages and DCs have been regarded as distinct cell types, with macrophages considered a part of the innate immune system, whilst DCs are antigen presenting cells that regulate immune effector cells. However, macrophages and DCs in the kidney and elsewhere frequently exhibit overlapping functional properties. In addition, the cells of the renal mononuclear phagocytic system can simultaneously express cell surface markers traditionally associated with either macrophages or DCs. Thus a growing body of researchers believe that macrophages and DCs should be regarded as cells that exist across a common spectrum. Thirdly, DCs can undergo a functional switch depending on the stage of disease. The precise mechanism controlling this process and how this contributes to the progression of renal disease is still unclear. Fourthly, the differences between human and mouse DC subset markers have added to the complexity, making it difficult to extrapolate information from experimental models to human kidney disease.
Conclusion
There have been a number of recent discoveries in the field of DC research which are uncovering the role of DCs in renal inflammation. Much has been learnt about DCs in experimental models of disease. However, our knowledge about the role of DCs in human kidney disease is still limited. Therefore, finding how to reliably extrapolate knowledge from experimental models to human kidney disease is a major challenge which must be tackled on the long road towards clinical translation.
Abbreviations
- AKI:
-
Acute kidney injury
- APCs:
-
Antigen presenting cells
- CNT:
-
Cisplatin nephrotoxicity
- CTL:
-
Cytotoxic T cells
- CXCL16:
-
Chemokine (C-X-C motif) ligand 16
- DAMPS:
-
Damage-associated molecular patterns
- DC:
-
Dendritic cell
- DT:
-
Diphtheria toxin
- HMGB-1:
-
High-mobility group box 1
- ICOS-L:
-
Inducible T cell co-stimulator ligand
- IFN:
-
Interferon
- IFN RF:
-
Interferon regulatory factor
- IL:
-
Interleukin
- iNKT:
-
Invariant natural killer T
- IRI:
-
Ischemia reperfusion injury
- MAMPs:
-
Microbe-associated molecular patterns
- MAPK:
-
Mitogen-activated protein kinase
- mDCs:
-
Myeloid DCs
- MSC:
-
Mesenchymal stem cells
- NKT:
-
Natural killer T
- NLRP3:
-
NOD-like receptor family, pyrin domain containing 3
- NOH:
-
Nephrin-OVA-HEL mice
- NTN:
-
Nephrotoxic nephritis
- PRRs:
-
Pattern-recognition receptors
- RLN:
-
Renal lymph node
- SIGIRR:
-
Single Ig/IL-1–related receptor
- Th:
-
T helper cells
- TLRs:
-
Toll-like receptors
- TNF-α:
-
Tumour necrosis factor alpha
- UUO:
-
Unilateral ureteral obstruction
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
John R, Nelson PJ (2007) Dendritic cells in the kidney. J Am Soc Nephrol 18(10):2628–2635
Rogers NM et al (2009) Review article: kidney dendritic cells: their role in homeostasis, inflammation and transplantation. Nephrology (Carlton) 14(7):625–635
Kushwah R, Hu J (2011) Complexity of dendritic cell subsets and their function in the host immune system. Immunology 133(4):409–419
Ferenbach D, Hughes J (2008) Macrophages and dendritic cells: what is the difference? Kidney Int 74(1):5–7
Cao Q et al (2014) Renal F4/80 + CD11c + mononuclear phagocytes display phenotypic and functional characteristics of macrophages in health and in adriamycin nephropathy. J Am Soc Nephrol. doi: 10.1681/ASN.2013121336
Lindenmeyer M et al (2011) Dendritic cells in experimental renal inflammation—Part I. Nephron Exp Nephrol 119(4):e83–e90
Soos TJ et al (2006) CX3CR1 + interstitial dendritic cells form a contiguous network throughout the entire kidney. Kidney Int 70(3):591–596
Steinman RM, Idoyaga J (2010) Features of the dendritic cell lineage. Immunol Rev 234(1):5–17
Lukacs-Kornek V et al (2008) The kidney-renal lymph node-system contributes to cross-tolerance against innocuous circulating antigen. J Immunol 180(2):706–715
Dong X et al (2007) Resident dendritic cells are the predominant TNF-secreting cell in early renal ischemia-reperfusion injury. Kidney Int 71(7):619–628
•• Hochheiser K et al (2011) Kidney Dendritic Cells Become Pathogenic during Crescentic Glomerulonephritis with Proteinuria. J Am Soc Nephrol 22(2): 306–16. This is the first study showing functional switch of DCs from an early protective to a later pro-inflammatory phenotype during the course of NTN
Tucci M et al (2009) Oversecretion of cytokines and chemokines in lupus nephritis is regulated by intraparenchymal dendritic cells: a review. Ann N Y Acad Sci 1173:449–457
Lee H et al (2010) CD4 + CD25 + regulatory T cells attenuate cisplatin-induced nephrotoxicity in mice. Kidney Int 78(11):1100–1109
Scholz J et al (2008) Renal dendritic cells stimulate IL-10 production and attenuate nephrotoxic nephritis. J Am Soc Nephrol 19(3):527–537
Liu Y, Shi G (2014) Role of G protein-coupled receptors in control of dendritic cell migration. Biomed Res Int 2014:738253
Teteris SA, Engel DR, Kurts C (2011) Homeostatic and pathogenic role of renal dendritic cells. Kidney Int 80(2):139–145
•• Hochheiser K et al (2013) Exclusive CX3CR1 dependence of kidney DCs impacts glomerulonephritis progression. J Clin Invest 123(10): 4242–54. This study identified CX3CR1 as a kidney specific chemokine receptor expressed by renal DCs which may offer a more specific target in treating progressive renal disease
Shen H, Kreisel D, Goldstein DR (2013) Processes of sterile inflammation. J Immunol 191(6):2857–2863
Rosin DL, Okusa MD (2011) Dangers within: dAMP responses to damage and cell death in kidney disease. J Am Soc Nephrol 22(3):416–425
Panzer U et al (2007) Chemokine receptor CXCR3 mediates T cell recruitment and tissue injury in nephrotoxic nephritis in mice. J Am Soc Nephrol 18(7):2071–2084
Heymann F et al (2009) Kidney dendritic cell activation is required for progression of renal disease in a mouse model of glomerular injury. J Clin Invest 119(5):1286–1297
Panzer U, Kurts C (2010) T cell cross-talk with kidney dendritic cells in glomerulonephritis. J Mol Med (Berl) 88(1):19–26
Noessner E et al (2011) Dendritic cells in human renal inflammation—Part II. Nephron Exp Nephrol 119(4):e91–e98
Friedewald JJ, Rabb H (2004) Inflammatory cells in ischemic acute renal failure. Kidney Int 66(2):486–491
Kinsey GR, Li L, Okusa MD (2008) Inflammation in acute kidney injury. Nephron Exp Nephrol 109(4):e102–e107
Li L, Okusa MD (2010) Macrophages, dendritic cells, and kidney ischemia-reperfusion injury. Semin Nephrol 30(3):268–277
Schlichting CL, Schareck WD, Weis M (2006) Renal ischemia-reperfusion injury: new implications of dendritic cell-endothelial cell interactions. Transplant Proc 38(3):670–673
Guo G et al (1999) Role of TNFR1 and TNFR2 receptors in tubulointerstitial fibrosis of obstructive nephropathy. Am J Physiol 277(5 Pt 2):F766–F772
Misseri R et al (2005) TNF-alpha mediates obstruction-induced renal tubular cell apoptosis and proapoptotic signaling. Am J Physiol Renal Physiol 288(2):F406–F411
Donnahoo KK et al (1999) Review article: the role of tumor necrosis factor in renal ischemia-reperfusion injury. J Urol 162(1):196–203
Leemans JC et al (2005) Renal-associated TLR2 mediates ischemia/reperfusion injury in the kidney. J Clin Invest 115(10):2894–2903
Allam R et al (2012) Histones from dying renal cells aggravate kidney injury via TLR2 and TLR4. J Am Soc Nephrol 23(8):1375–1388
Wu H et al (2007) TLR4 activation mediates kidney ischemia/reperfusion injury. J Clin Invest 117(10):2847–2859
Kurts C et al (2013) The immune system and kidney disease: basic concepts and clinical implications. Nat Rev Immunol 13(10):738–753
Wu H et al (2010) HMGB1 contributes to kidney ischemia reperfusion injury. J Am Soc Nephrol 21(11):1878–1890
• Li L et al (2012) Dendritic cells tolerized with adenosine A(2)AR agonist attenuate acute kidney injury. J Clin Invest 122(11): 3931–42. This study showed that tolerized DCs, induced by A2AR agonist ATL313, suppress NKT cell activation in vivo and prevent NKT cell-mediated innate immune activation in IRI. This provides a unique cell-based strategy to attenuate IRI
Dong X et al (2005) Antigen presentation by dendritic cells in renal lymph nodes is linked to systemic and local injury to the kidney. Kidney Int 68(3):1096–1108
Lech M et al (2009) Resident dendritic cells prevent postischemic acute renal failure by help of single Ig IL-1 receptor-related protein. J Immunol 183(6):4109–4118
Lassen S et al (2010) Ischemia reperfusion induces IFN regulatory factor 4 in renal dendritic cells, which suppresses postischemic inflammation and prevents acute renal failure. J Immunol 185(3):1976–1983
Cho WY et al (2010) The role of Tregs and CD11c(+) macrophages/dendritic cells in ischemic preconditioning of the kidney. Kidney Int 78(10):981–992
Togel F et al (2007) Vasculotropic, paracrine actions of infused mesenchymal stem cells are important to the recovery from acute kidney injury. Am J Physiol Renal Physiol 292(5):F1626–F1635
• Kim MG et al (2013) CD11c(+) cells partially mediate the renoprotective effect induced by bone marrow-derived mesenchymal stem cells. PLoS One 8(8): e72544. This is the first study demonstrating the renoprotective effect of MSCs is partially mediated by their direct interaction with CD11c + cells, which is in part dependent on IL-10
• Kim MG et al (2010) Depletion of kidney CD11c + F4/80 + cells impairs the recovery process in ischaemia/reperfusion-induced acute kidney injury. Nephrol Dial Transplant 25(9): 2908–21. This is the first study showing DCs change from a pro-inflammatory to an anti-inflammatory phenotype during recovery phase of IRI
Dong X et al (2008) Dendritic cells facilitate accumulation of IL-17 T cells in the kidney following acute renal obstruction. Kidney Int 74(10):1294–1309
Kitamoto K et al (2009) Effects of liposome clodronate on renal leukocyte populations and renal fibrosis in murine obstructive nephropathy. J Pharmacol Sci 111(3):285–292
Machida Y et al (2010) Renal fibrosis in murine obstructive nephropathy is attenuated by depletion of monocyte lineage, not dendritic cells. J Pharmacol Sci 114(4):464–473
Snelgrove SL et al (2012) Renal dendritic cells adopt a pro-inflammatory phenotype in obstructive uropathy to activate T cells but do not directly contribute to fibrosis. Am J Pathol 180(1):91–103
Tadagavadi RK, Reeves WB (2010) Endogenous IL-10 attenuates cisplatin nephrotoxicity: role of dendritic cells. J Immunol 185(8):4904–4911
Tadagavadi RK, Reeves WB (2010) Renal dendritic cells ameliorate nephrotoxic acute kidney injury. J Am Soc Nephrol 21(1):53–63
• Mulay SR et al (2013) Calcium oxalate crystals induce renal inflammation by NLRP3-mediated IL-1beta secretion. J Clin Invest 123(1): 236–46. This study demonstrated DCs contribute to inflammation in crystal nephropathy through activation of NLRP3 inflammasome cascade and secretion of the pro-inflammatory cytokine IL-1β. IL-1β blockade may prevent renal damage in nephrocalcinosis
Tipping PG, Holdsworth SR (2006) T cells in crescentic glomerulonephritis. J Am Soc Nephrol 17(5):1253–1263
Kruger T et al (2004) Identification and functional characterization of dendritic cells in the healthy murine kidney and in experimental glomerulonephritis. J Am Soc Nephrol 15(3):613–621
Kitching AR et al (2002) Interleukin-10 inhibits experimental mesangial proliferative glomerulonephritis. Clin Exp Immunol 128(1):36–43
Yang SH et al (2008) NKT cells inhibit the development of experimental crescentic glomerulonephritis. J Am Soc Nephrol 19(9):1663–1671
Riedel JH et al (2012) Immature renal dendritic cells recruit regulatory CXCR6(+) invariant natural killer T cells to attenuate crescentic GN. J Am Soc Nephrol 23(12):1987–2000
Kim AH, Markiewicz MA, Shaw AS (2009) New roles revealed for T cells and DCs in glomerulonephritis. J Clin Invest 119(5):1074–1076
Hochheiser K, Tittel A, Kurts C (2011) Kidney dendritic cells in acute and chronic renal disease. Int J Exp Pathol 92(3):193–201
Iwata Y et al (2009) Dendritic cells contribute to autoimmune kidney injury in MRL-Faslpr mice. J Rheumatol 36(2):306–314
• Castellano G et al (2010) Infiltrating dendritic cells contribute to local synthesis of C1q in murine and human lupus nephritis. Mol Immunol 47(11–12): 2129–37. This study demonstrated DCs exacerbate lupus nephritis by contributing to local synthesis of C1q. This may represent a potential new target for future treatment of lupus nephritis
Woltman AM et al (2007) Quantification of dendritic cell subsets in human renal tissue under normal and pathological conditions. Kidney Int 71(10):1001–1008
Segerer S et al (2008) Compartment specific expression of dendritic cell markers in human glomerulonephritis. Kidney Int 74(1):37–46
Fiore N et al (2008) Immature myeloid and plasmacytoid dendritic cells infiltrate renal tubulointerstitium in patients with lupus nephritis. Mol Immunol 45(1):259–265
•• Kassianos AJ et al (2013) Increased tubulointerstitial recruitment of human CD141(hi) CLEC9A(+) and CD1c(+) myeloid dendritic cell subsets in renal fibrosis and chronic kidney disease. Am J Physiol Renal Physiol 305(10): F1391–401. This is the first study using novel methods for the extraction of DC subsets from human kidney biopsy tissue for multicolor phenotypic and quantitative characterization. It identified myeloid DCs as a key producer of pro-fibrotic cytokine TGF-β
Tucci M et al (2008) Glomerular accumulation of plasmacytoid dendritic cells in active lupus nephritis: role of interleukin-18. Arthritis Rheum 58(1):251–262
Wilde B et al (2009) Dendritic cells in renal biopsies of patients with ANCA-associated vasculitis. Nephrol Dial Transplant 24(7):2151–2156
Zheng D et al (2012) Lipopolysaccharide-pretreated plasmacytoid dendritic cells ameliorate experimental chronic kidney disease. Kidney Int 81(9):892–902
Zheng D et al (2014) Adoptive transfer of bone marrow dendritic cells failed to localize in the renal cortex and to improve renal injury in adriamycin nephropathy. Nephron Exp Nephrol 126(1):8–15
Acknowledgments
This work was supported by the National Health & Medical Research Council of Australia (NHMRC, Grants 457345 and 632665 to Yiping Wang & David Harris).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Chen, T., Cao, Q., Wang, Y. et al. The Role of Dendritic Cells in Renal Inflammation. Curr Pathobiol Rep 2, 225–234 (2014). https://doi.org/10.1007/s40139-014-0059-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40139-014-0059-6