
Abstract
Purpose of Review
This review sought to assess the extent and causes of suboptimal healthcare outcomes for chronic variable immunodeficiency (CVID).
Recent Findings
Significant improvements in diagnostic technology and treatment protocols over time were found, leading to reduced morbidity and mortality for those accessing therapies. Treatments continue to be largely non-curative with financing (mainly insurance coverage) an obstacle. Symptom recognition by primary care practitioners (PCP) remains a gating factor to treatment and a widespread and persistent barrier to optimal health outcomes.
Summary
CVID is a subtype of primary immunodeficiency (PIDD) associated with under-diagnosis. It has emerged as a health issue more prevalent than historically known. No symptom-recognition framework for early detection of CVID has been generally accepted; those proposed for primary immunodeficiencies have shown low sensitivity, low specificity or both. Positive trends in cases diagnosed have been aided by awareness campaigns and international collaborations. However, treatments for CVID will not realize full potential without effective, accepted frameworks for timely identification in the clinic.
Similar content being viewed by others
Introduction
The history of scientific understanding of primary immunodeficiency (PIDD) is relatively short but emerging rapidly. It was not until 1952 that primary immunodeficiency was first recognized as a disease. Before the widespread use of antibiotics, mortality from infections was common, rarely prompting autopsies [1, 2]. It was only afterwards that chronic or recurrent infections became distinguishable, encouraging further scientific investigation [1]. In 1970, the World Health Organization convened the first international committee tasked with forming consensus around PIDD class and subclass definitions, best practices for diagnosis and treatment, and establishment of patient registries [3]. The committee, now under the International Union of Immunological Societies and named the Inborn Errors of Immunity Committee, meets and publishes an updated, growing classification list every 2 years.
From the initial broad “humoral” and “cellular” descriptions prior to 1970, 14 distinct subclasses resulted from the first World Health Organization meeting [3]. As an example, it was at this 1970 meeting that the term chronic variable immunodeficiency (CVID) was first described as a hypogammaglobulinemia “diagnosis of exclusion” that should not be made before 4 years of age [4•]. By 2003, the number of identified primary immunodeficiencies grew to 80, then to 240 by 2015. A 2017 Inborn Errors of Immunity Committee report described 330 primary immunodeficiencies with 320 specific, associated genetic defects [5]. Just months later, its updated February 2018 list included 355 distinct primary immunodeficiencies within 39 subcategories and 341 associated genetic defects [6]. Since 2012, the majority of new PIDD-associated genetic defects have been discovered using next generation sequencing technology, and the pace of discovery is expected to continue as sequencing becomes more accessible [6].
Understanding of the scope of PIDD as a healthcare issue has grown in recent years. Prevalence, in total and by subtype, was found to be far from definitively established, but estimates increased dramatically over time, aided by diagnostic capabilities, data collection, and awareness campaigns. Inter-study comparisons were found to be of limited use due to variability in time, sample size, geographic and demographic scope, disease classification, methodology, and other factors. Prevalence research could not adequately adjust for the existence of large, known undiagnosed populations [4•]. The Immune Deficiency Foundation sponsored the first survey in 1995 of self-identified, diagnosed PIDD patients and physicians who treat PIDD. While the primary objective was to gather information regarding confirmed cases, an estimate of 50,000 U.S. PIDD cases was formulated, equating to prevalence of 1 in 5300 [7].
Joshi et al. analyzed ICD-9 diagnostic codes collected by the Mayo Clinic for Olmsted County, Minnesota, over a 31-year period from 1976 through 2006. This study reported an incidence of 1 in 9700 person-years during the 5-year period ending December 2006, representing a 4.3-fold increase compared to an earlier 5-year period ending December 1980 (1 in 41,700) [8]. Another ICD-9 code-based study by Kobrynski et al. from a national patient database (MarketScan) estimated prevalence as 1 in 2000 among privately insured and 1 in 2400 among Medicaid recipients in 2007. These figures represented 30 and 42% increases over 2001 (1 in 2600 and 3400, respectively) [9].
A national random sample of 10,005 households conducted in 2005 found 23 members of 18 households reporting confirmed diagnoses of PIDD, equating to prevalence of 1 in 1200, more than 8-fold higher than seen by Joshi et al. Extrapolating to the 2005 population, this study’s authors estimated 250,000 cases in the USA (152,000–361,000, 95% confidence interval), 5-fold greater than the above-described Immune Deficiency Foundation (1995) estimate, pushing PIDD over the U.S. National Institutes of Health 200,000 “rare disease” threshold [2]. Subsequently, 250,000 has been frequently cited in published literature but the National Institutes of Health website reports the figure as 500,000, a number that includes a large, typically asymptomatic selective IgA deficiency population [10]. Globally, Bousfiha et al. reviewed multiple studies and applied rates from developed healthcare economies to those countries assumed to underreport PIDD. They estimated that only 10% of primary immunodeficiencies had been diagnosed, with as many as 6,000,000 cases worldwide [11].
Undiagnosed PIDD has not been credibly estimated, but is widely referred to in published literature as a large population. The Jeffrey Modell Foundation, which funds PIDD research, testing, awareness, and treatment, has developed a global network of diagnostic and research centers and expert physicians at academic hospitals [12]. A survey of their network for the 2-year period from 2013 to 2015 showed an increase of 77.9% in U.S. PIDD referrals (25.5% globally), a 19.4% increase in new diagnoses (8.5% globally), and a 30% increase in patients evaluated and tracked (13.4% globally) [13••]. PIDD subtypes were not broken out in this report. Significant intra-study temporal increases of primary immunodeficiencies have been reported which provide further evidence of historic under-diagnosis [8, 9, 13••]. In fact, Joshi et al. postulated that the increase between the two time points measured in their study could be attributable to improved diagnostic technology and physician awareness [8]. Reviewing ICD-9 codes from the largest pediatric healthcare database in the USA, the Kids’ Inpatient Database, from 2003 through 2012, identified 26,794 (1 in 1083) hospitalizations as PIDD related. The incidence of PIDD admissions nearly doubled during that period from 1 in 1501 in 2003 to 1 in 789 in 2012 [14].
Studies have attempted to quantify undiagnosed PIDD in specific populations with varying success. A 2004 study aimed to identify minorities—generally underrepresented in PIDD patient registries—with undiagnosed PIDD. PIDD-related ICD-9 codes for admissions to one urban hospital between 1995 and 2001 were analyzed. Of this patient pool, 533 (or 0.4%) were identified using a scoring system. Fifty-nine patients of those identified were randomly selected for diagnostic tests. Seventeen (29%) were ultimately diagnosed with PIDD, 13 (22%) with secondary immunodeficiencies (most frequently related to sickle cell anemia), and 29 (49%) had no detectable immune deficiency. For those diagnosed with PIDD, the most common infections were pneumonia and bronchiectasis [15]. While the specificity of the scoring system was low, this work succeeded in illustrating significant under-diagnosis of PIDD in this population. More recent efforts to identify undiagnosed primary immunodeficiencies are discussed below.
Owing in part to technological advances, the pace of discovery of biologic drivers and identifiable PIDD phenotypes has accelerated over the past decade. The history of acute primary immunodeficiencies, such as severe combined immunodeficiency (SCID), popularly referred to as “bubble boy syndrome,” hints at the underestimation of the prevalence of less severe primary immunodeficiencies. Untreated SCID often leads to death within the first year of life, but availability of treatments hastened its inclusion into neonatal blood test panels in all 50 states using T cell receptor excision circle assays [16]. Consequently, reported SCID incidence nearly doubled to 1 in 58,000 from 1 in 100,000 [17]. Other primary immunodeficiencies are not yet included in routine blood tests.
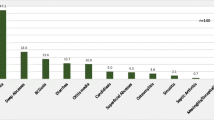
Compared to SCID, CVID is reported to have an incidence of as high as 1 in 10,000. It results from hypogammaglobulinemia, an inability of the body’s B cells to produce sufficient numbers of functional circulating immunoglobulins (Ig) or antibodies. Without adequate Ig, those with CVID often suffer recurrent and chronic bacterial infections, mostly of the upper and lower respiratory tracts, but occasionally of the gastrointestinal tract as well. CVID is considered a “diagnosis of exclusion,” a basket for which no molecular or genetic markers can be attributable to a defined antibody deficiency [4•, 18]. It is the most prevalent (27%) type of primary antibody deficiency (also known as a B cell deficiency or humoral deficiency) and primary antibody deficiencies are the largest subset (57%) of PIDD. Other major PIDD subsets involve both B and T cells (20%), phagocytes (4%), and complement (2%) (Fig. 1) [19•]. Secondary (or “acquired”) immune deficiency is more common than primary and can be caused by many indirect factors, such as poor nutrition, infections (e.g., AIDS), corticosteroids, cancer, and treatment with chemotherapy [20]. Finally, a myriad of common environmental factors (e.g., frequent exposure to allergens or children with colds in a daycare setting, etc.) that challenge the immune system cause symptoms that primary care practitioners (PCPs) routinely see in the clinic and are unrelated to any immune system defect [21].

Classification and type of immunodeficiencies reported by Jeffrey Modell Diagnostic/Referral Centers adapted from Modell et al. [19•]
Similar to PIDD discussed above, a wide range of CVID prevalence estimates were found in published literature. The most commonly referenced was 1 in 25,000, with ranges from 1 in 10,000 to 1 in 100,000. All are based on flawed data and methods [4•, 6, 22,23,24]. CVID represented 15.4% of primary immunodeficiencies according to a Jeffrey Modell Foundation survey of its network centers (Fig. 1) [19•]. This proportion would imply a prevalence of 1 in 7700 if applied to Boyle & Buckley’s findings, or 1 in 12,800–15,400 to Kobrynski et al.’s [2, 9]. CVID represented much higher percentages of primary immunodeficiencies in the European Society for Immunodeficiencies (ESID) and U.S. Immunodeficiency Network registries (21 and 30%, respectively), which would imply a CVID prevalence of 1 in approximately 4000–5700 applied to Boyle & Buckley’s findings or 1 in approximately 6700–11,400 if applied to Kobrynski et al.’s [2, 4•, 9, 19•].
Specific Aims
The aim of this critical analysis is to assess barriers to optimal healthcare outcomes in the population with CVID. Specifically, barriers and benefits to reducing diagnosis delays will be discussed. Current standards and practices for recognizing, diagnosing, and treating CVID will be reviewed in this context.
Methods
A systematic literature review was conducted to include articles published in English, limited to the period January 1, 2004 through February 15, 2019. Selected literature published prior to 2004 was referenced for historic context. Databases used were Pubmed, Scopus, Google Scholar, and Health Information @Himmelfarb. Keywords included CVID, immunoglobulin, infection, diagnosis, treatment, and delay, yielding over 3000 results. Titles were scanned and abstracts read when titles appeared relevant. Approximately 125 articles were identified as relevant. Additional resources included websites and publications of the Immune Deficiency Foundation and Jeffrey Modell Foundation.
Results
Confounding Factors to Timely Diagnosis
Several factors were found to confound early diagnosis of CVID. First, unlike SCID, neonatal CVID testing was not recommended [25]. While CVID has been seen in children as young as two, immaturity of the immune system at that age makes current test specificity low [4•]. Early studies drawing on data from small registries indicated CVID diagnoses clustered within two age ranges—6 to 10 and 26 to 40, but later studies of larger cohorts showed a continuous curve [24, 26]. Symptom onset, unlike most primary immunodeficiencies, has been seen over a very broad age range, from the first to the ninth decade of life; for this reason the International Consensus Document for CVID recommended that “clinicians must maintain an index of suspicion for antibody deficiency in patients of all ages” [4•, 27].
Second, significant heterogeneity within CVID was found. Three types of immunoglobulins, or “isotypes”—IgG, IgA, and IgM—are involved in helping the body fight infections by identifying and attaching to invading antigens, rendering them harmless or marking them for destruction by other immune system components. The levels of each in CVID have been found to be highly variable [28]. Considerable effort by international immunology organizations has produced consensus CVID definitions. While there are five classes of Ig—IgG, IgA, IgM, IgD, and IgE—the current definition stipulated that IgG and either IgA or IgM, or both, be at least 2 standard deviations under age and ethnicity-adjusted means [4•, 24]. Under that threshold, however, considerable variability has been measured [29]. Conversely, patients who were seen presenting with CVID-defining symptoms, the requisite IgG deficiency, but normal IgA or IgM levels, have been categorized as “CVID-like” [30]. Some symptomatic patients have been assessed with normal total IgG but deficient in one or more subclasses (IgG1, IgG2, IgG3, or IgG4) linked to multiple phenotypes. Other biomarkers, such as percent of isotype-switched memory B cells, have also been determined to influence phenotype [28].
Third, genetic and other potential biological drivers of CVID were found to be largely unknown, especially as compared to many other primary immunodeficiencies, although this was evolving. There are limited, but growing, numbers of single gene defects discovered to date [31]. While a small, recent study of 36 pediatric CVID patients identified 15 to 24% with monogenic drivers, a lower 2 to 10% is the range often cited for all CVID age groups. Monogenic defects were found to be a larger percentage of pediatric-onset CVID and somewhat more likely to be hereditary (as opposed to sporadic) [4•, 32]. Review of family history was described as an important physician tool to assess risk for many primary immunodeficiencies, but the International Consensus Document for CVID reported only 5 to 25% of cases are familial [4•].
The majority of diagnosed CVIDs are found to be of sporadic origin and were attributed to complex interactions between multiple genetic, and possibly epigenetic, factors. Studies are ongoing to understand how sporadic CVID may be triggered, including environmental factors that influence DNA methylation (a mechanism of epigenetics) when genetic predisposition exists [33]. There are likely many conditions causing predisposition, one example being selective IgA deficiencies progressing to CVID [34].
Fourth, the large majority of CVIDs have been reported as presenting in the clinic with repeat, common bacterial infections, such as sinus, bronchial, gastrointestinal, and ear infections [24]. In other words, a malfunctioning immune system with CVID often manifests in the clinic as well-described infections typically not caused by any immune system defect.
Those with PIDD rely predominantly on general healthcare practitioners to initiate appropriate healthcare workup with subsequent interventions. For non-SCID PIDD, such as CVID, the prototypical healthcare intervention cycle in the USA would flow as follows:
-
1.
PCP recognizes potential symptoms of immune deficiency, and may order common tests to guide next steps;
-
2.
PCP provides referral to a specialist, such as an allergist and/or immunologist;
-
3.
Specialist orders advanced blood tests;
-
4.
Specialist makes a diagnosis of a specific PIDD subclass (e.g., CVID);
-
5.
Specialist recommends an appropriate treatment protocol; and
-
6.
PCP and specialist monitor patient symptoms, coordinate treatment, and adjust protocols as necessary for maximum health benefit [21].
In the USA, PCPs are typically the gatekeepers to appropriate treatment for people with CVID; symptom recognition by PCPs is, therefore, an important gating factor.
Impact of Delay in Diagnosis of CVID
Approximately 80% of CVID diagnoses were reported as preceded by one or more incidences of pneumonia [35]. Permanent, structural lung damage, including bronchiectasis, was described as a common consequence of chronic respiratory tract infections and repeated pneumonia [36]. Severe, opportunistic infections were also seen including bacterial meningitis and sepsis. A large European multicenter study examined 2700 cases in 23 countries and reported non-infectious complications of CVID including autoimmune disease (25%), splenomegaly (24%), gastrointestinal disease/malabsorption (9.9%), granuloma (9.1%), and cancer (14.1%). More than one quarter of the patients included (26.8%) had developed chronic lung disease in the form of bronchiectasis. This study estimated the individual burden of CVID using disability life years (DALY) as 36,785 per 100,000, which was higher than depression disorders, diabetes mellitus, or chronic obstructive pulmonary disease [37•]. DALY is a standardized method of calculating the burden of a specific condition that incorporates data on years of life lost (YLL) and years living with a disability (YLD) linked to that condition. Similar comorbidities were seen consistently across most CVID studies [25, 38].
Delays from onset of symptoms to CVID diagnosis were analyzed in several large-cohort studies over the past decade and seen to be widespread and persistent. An average delay of 7.46 years (ranging from 0 to 60 years) was calculated among 338 patients in the European CVID registry. In this group, delay length was inversely related to age of CVID onset [26]. An Italian study of 224 CVID patients revealed an average delay of 8.9 years [39]. Approximately 20% of adults ultimately diagnosed with CVID were able to identify disease symptoms from childhood that had gone undiagnosed [40]. Non-infectious complications, while less common and often severe, did not necessarily raise PCP index of suspicion for CVID. For example, a Norwegian study found that patients with non-infectious complications had a median time to diagnosis of 17 years versus 10 for the infection-only cohort. On average, granulomatous disease (a lymphoproliferative disease) had been present 19 years prior to diagnosis, alopecia (an autoimmune disease) 25 years, and gastrointestinal diseases 13.5 years [41].
Diagnostic delay of CVID was strongly associated with complications and comorbidities in several studies. A German pediatric study reported multiple severe and chronic infections during an average 5.8-year delay to diagnosis and no serious infections after initiation of treatment. This study also showed association between delay in diagnosis and bronchiectasis [24]. The 2700-case CVID study by Odnoletkova et al. described above revealed diagnostic delay associated with bronchiectasis, solid tumors, and enteropathy [37•]. Each year of delay was associated with 1.4% increased mortality in a pan-European cohort of 2212 patients [42].
The Jeffrey Modell Foundation asked its network physicians to compile statistics for the year prior to and year after diagnoses of their patients. Their analysis, which incorporated all primary immunodeficiencies excluding SCID, showed post-diagnosis reductions of 84% in hospitalization days, 83% in physician/emergency room visits, 56% in days on antibiotics, and 74% in work or school days missed (Table 4) [13••]. Improvements can be attributed to effective treatments, including immunoglobulin replacement therapy (IgRT) and appropriate use of antibiotics, described below.
Identification of Undiagnosed Patients
Several frameworks proposed to improve PCP identification of primary immunodeficiencies were reviewed. The Jeffrey Modell Foundation and Immune Deficiency Foundation were found to be leaders in ongoing efforts to raise awareness among physicians and the public, especially in the USA. The Jeffrey Modell Foundation developed a global awareness campaign around its “Ten Warning Signs of Primary Immunodeficiency” (“Ten Warning Signs”) starting 1993. The Ten Warning Signs urge follow-up with healthcare professionals if two or more of the following are present (Table 1).
The Jeffrey Modell Foundation also developed and distributed to provider networks and insurers, free of charge, the SPIRIT Analyzer (Software for Primary Immunodeficiency Recognition, Intervention, and Tracking). SPIRIT Analyzer matches patient records to Ten Warning Sign-related ICD-10 codes in patient databases. A point scoring system assigns each patient to high, medium, and low, or zero PIDD risk. A test on a 2,000,000 patient sample from a large dataset (IMS LifeLink Health Plan Claims Database) identified over 1 in 1685 as high and 1 in 680 as moderate risk (combined, 1 in 485) [43]. However, no analysis on sensitivity and specificity for SPIRIT Analyzer could be found that validated the effectiveness of this program for real-world application.
The Cleveland Clinic tested the sensitivity of the Ten Warning Signs by retrospective analysis of electronic health records for the years 2006 through 2016. They identified 287 adults with confirmed PIDD (77% of whom had CVID) and 115 pediatric patients. Detailed case review revealed that only 45% of these adults and 64% of children had two or more of the warning signs [44]. In other words, 55 and 36% respectively, would have been missed if the Ten Warning Signs had been the sole identification criterion. A 2011 study in Northern England determined that only 62% of confirmed PIDD cases exhibited two or more of the warning signs; conversely, 38% had less than two. The authors of this study described shortcomings of the Ten Warning Signs, including that it does not address non-infectious complications. They advocated instead for resource allocation to training and laboratory diagnostics [45]. A University of Manchester study of 430 pediatric PIDD patients and 133 controls also showed low sensitivity and specificity. These authors proposed a simple decision tree framework for PCPs that hinges on past receipt of intravenous antibiotics or antifungals for sepsis [46].
Two German groups, the German Patients’ Organisation for Primary Immunodeficiencies (DSAI) and the Association of the Scientific Medical Societies in Germany (AWMF), attempted to improve on the sensitivity and specificity of the Ten Warning Signs. A third group from Heinrich Heine University, Dusseldorf (Dusseldorf), tested the DSAI and AWMF frameworks, along with two of its own, on a cohort of 210 PIDD cases. The Jeffrey Modell Foundation’s Ten Warning Signs were found to have the highest specificity but lowest sensitivity. The highest sensitivity (75%) was seen with Dusseldorf’s maximum negative predictive value (NPV) approach, but specificity was only 51% (Table 2) [47].
ESID developed a significantly more thorough framework for PCPs. Eight distinct clinical presentation categories were established, with corresponding columns for each, including (1) “special features” to look out for; (2) relevant pathogens; (3) potential alternative (non-PIDD) causes of clinical presentation; and (4) references for each to distinct step-by-step diagnostic protocols. Unlike the Ten Warning Signs and similar lists, the ESID framework did not suggest specific numbers of infections requiring follow-up. The authors described their approach in practical terms from a cost-benefit perspective, calling for immediate laboratory tests if severe, potentially life-threatening primary immunodeficiencies were possible (even if not probable), and phased-in examination and tests for cases unlikely to pose immediate, high risk [48]. The complexity of the ESID framework appeared to present a potential hurdle to widespread adoption in the PCP setting.
The above PCP recognition frameworks were developed by specialists (such as immunologists) for whom PIDD would likely be a priority. A different perspective on appropriate index of suspicion was observed in widely used publications for general PCP practice. For example, Nelson Pediatric Symptom-Based Diagnosis guides that immunodeficiency is fifth of five categories of consideration when patients present with recurrent, common infections. Category 1 is “the patient who is probably healthy,” guiding that children with no immune defects, especially in large families or day care, may have six to ten upper respiratory infections or gastroenteritis episodes per year. Six infection-related criteria that would individually, or combined, warrant further investigation were listed as follows: (1) more than two systemic bacterial infections (sepsis, meningitis, osteomyelitis); (2) two or more serious respiratory infections (pneumonia, sinusitis) or bacterial infections (cellulitis, draining otitis media, lymphadenitis) per year; (3) the presence of an infection at an unusual site (hepatic or brain abscess); (4) infections with unusual pathogens (Aspergillus pneumonia, disseminated candidiasis, or infection with Serratia marcescens, Nocardia species, or Burkholderia cepacia); (5) infections of unusual severity; and (6) recurrent mycobacterial infections or invasive infections with atypical mycobacteria [21]. This framework places more emphasis on serious infections, a focus that could miss the majority of recurrent, common infections (e.g., frequent or chronic sinus and upper respiratory tract infections) characteristic of most CVIDs.
The diagnostic approach described in Nelson Pediatric Symptom-Based Diagnosis and other similar guidebooks was considered in the context of PCP training and awareness. The American Academy of Allergy, Asthma, & Immunology (AAAAI) Primary Immune Deficiency Committee and the Immune Deficiency Foundation collaborated on a mail-based survey of 4500 PCPs in 2009 to assess awareness, training, and practice strategies among family physicians. Twelve percent (528) were returned, 44% affirming at least one PIDD patient followed, and 56% not following any. Only 9% of PCPs followed one or more patient with CVID. PIDD was described as insufficiently covered in medical school, with 11% responding it was not covered at all and 76% just minimally. Only 12% reported adequate coverage. Responses regarding continuing education revealed gaps as well; 99% had not participated in PIDD-related education during the preceding 6 months but 66% were interested in doing so [49]. A similar 2008 survey reported 93% of responding pediatricians being unaware of the existence of published PIDD guidelines and 35% uncomfortable with PIDD symptom recognition [50]. Of a survey of 1880 PCPs, only 32% knew of one patient with PIDD within the past 5 years. Only 10% recalled receiving a copy of the Ten Warning Signs [20].
A positive sign that interest and awareness may be higher today than when the above surveys were taken could be AAAAI’s 2018 Diagnostic School in Primary Immunodeficiencies. This was the organization’s first ever such continuing education seminar for physicians looking to “effectively and appropriately harness diagnostic tools currently available in order to provide the best benefit to their patients” [51].
Technical Capabilities to Diagnose CVID
Technology did not appear to be an impediment given the existence of tests to confer diagnoses sufficient for directing treatment. Furthermore, guidelines for appropriate tests and interpretation of results are widely available [4•]. Relative to potential implications of delayed diagnosis described above, accessible, incremental tests may provide cost-effective means for PCPs to narrow their focus and ultimately determine if referral to a specialist would be appropriate. For example, sputum culture was cited as an initial, effective means of detecting presence of encapsulated bacteria associated with PIDD subtypes. Complete blood count with differential, serum Ig, complement, and antibody titers (to measure expected immune response from past vaccines) were considered effective tools for PCP initial screens [52]. Specialists were seen to rely on standard flow cytometry tests to measure a large and diverse number of cell types and cell surface proteins involved in the normal immune response. Standard immune cell function tests were used to gauge in vitro leukocyte responses to a number of stimuli, including antigens (e.g., tetanus and candida) and mitogens (e.g., pokeweed, phytohemagglutinin, and concanavalin A). These tests were described as highly effective in identifying T cell, B cell, phagocyte, complement, or other immune system defects, required by specialists to prescribe appropriate treatments. As mentioned, guidelines such as the International Consensus Document for CVID are readily available for interpretation of laboratory results with regard to disease definitions [4•].
In lieu of genetic markers for guidance, significant compensatory work was reviewed that successfully stratified CVID risk into homogenous clinical phenotypes. A 2012 study of three independent cohorts found meaningful associations between biomarkers and phenotype, as well as with morbidity and mortality, with the following four classifications: (1) no other disease-related complications (infections only); (2) autoimmune cytopenias; (3) polyclonal lymphoproliferation (including noninfective granuloma, lymphoid interstitial pneumonitis, and persistent unexplained lymphadenopathy); and (4) unexplained enteropathy. Ninety percent of CVID patients in the study were found to have symptoms relating to one phenotype only. Sixty-three percent to 78% fell within the “infections only” phenotype. Associations between biomarkers and risk were found, including IgG levels and cytopenia, IgA levels and lymphoproliferative disease, and both IgM and CD4 levels and heptomegaly. Mean overall survival was associated with classification (highest to lowest in classification number order) [18]. Risk stratification allows PCPs and specialists to be on the alert for specific symptoms in patients diagnosed with CVID that might require immediate attention and allow for improved family counseling after making the diagnosis.
Mining of growing databases have shown meaningful associations between multiple copy number variations and clinical phenotypes. One study identified copy number variations that could predict 16 distinct clinical CVID subgroups (including, among others, lymphoma, bronchiectasis, lymphadenopathy, granuloma, gastrointestinal enteropathy, malabsorption, splenectomy, cytopenias, and organ-specific autoimmunity) with 98.7% accuracy [53]. As the cost of next generation sequencing decreases and patient databases grow, more combinations of alleles could more accurately predict potential complications of CVID.
Effectiveness of Treatment Protocols for CVID
The most important advancement for CVID treatment described in published research has been Ig replacement therapy. IgRT has been nearly universally indicated for CVID [54]. Today, IgRT products are derived from purified plasma collected from thousands of healthy donors. Products include a diverse spectrum of antibodies to afford protection from infections [55]. Human immune globulin concentrate was available during World War II and IgRT was first used in 1952, but safer products (e.g., enhanced screening and reliable purification to eliminate pathogens) were required for the widespread approved use seen in the 1980s [55]. Manufacturing efficiency and product stability increased availability.
Initially administered via frequent, painful intramuscular injections, higher concentration Ig products can now be administered monthly intravenously or at shorter intervals subcutaneously. Subcutaneous Ig permitted home self-administration, with flexible dosing and scheduling. Recombinant hyaluronidase was recently incorporated into a subcutaneous product (rHuPH20) that temporarily breaks down the skin’s extracellular matrix, allowing less frequent, faster administration of larger Ig quantities. Improved patient adherence has in part been attributed to improved safety, efficacy, tolerability, and convenience [55, 56].
Early, low fixed IgRT doses showed mixed results, but the product enhancements described above facilitated research on dose optimization. A European quality of life study revealed 58% higher average number of days spent in hospital by CVID patients who were not receiving IgRT than those who were [57]. Titration to individualized, optimal trough Ig levels, referred to as the “biologic level,” has been reported to improve infection suppression significantly [55, 58, 59]. Review of dosing evolution—from early standard doses of 25 mg/kg (body weight), to 50 in the 1960s, then 200 to 500 during the 1980s, to as high as 1000 mg/kg today, provided essential context for comparison of efficacy studies over time. Recent studies using higher and/or biologic levels were seen to show stronger association with suppression of infections, including pneumonia [35].
IgRT was not the only therapy described as essential for optimal CVID and other primary antibody deficiency health outcomes. In fact, some antibody deficiencies do not benefit from IgRT at all. A review of commercially available products revealed that IgA and IgM, both of which have much shorter half-lives than IgG, are not material components of IgRT, making them ineffective for selective IgA or IgM deficiencies [60]. At the same time, the paucity of IgA and IgM in commercially available IgRT was reported to underlie continued susceptibility to certain types of infections for those with CVID. Secreted IgA2 (one of two types of IgA), for example, lines mucosal surfaces such as the respiratory and digestive tracts, directly neutralizing antigens there. IgA2 deficiency—often seen with CVID—has been associated with sinus and upper respiratory tract infections even after IgRT initiation, though on average at reduced rates [61]. Several studies reported pneumonia, bacterial meningitis, and sepsis rates dramatically reduced once biologic Ig levels were reached, but sinusitis, bronchiectasis, and splenomegaly did not [62].
Antibiotic therapy is an essential, ongoing component of treatment for breakthrough infections with CVID, regardless of IgRT use or prophylactic antibiotics. Though overuse of antibiotics is a growing concern, the benefits of prophylactic use have outweighed the risks for some [22, 25]. Prophylactic antibiotics have been shown to ameliorate chronic granulomatous disease, a potential complication of CVID, for example [63]. However, appropriate patient and family counseling is necessary when utilizing prophylactic antibiotics. One study found that patients on prophylactic antibiotics delayed seeking necessary high-dose prescriptions for breakthrough infections [64].
Recommendations for vaccinations need to be determined based on the specific type of CVID subtype in question. For example, IgG1 and IgG3 respond to certain vaccine protein antigens while IgG2 and IgG4 respond only to polysaccharide antigens. Although those treated with IgRT will have acquired many vaccine-induced antibodies from plasma donors, there appeared to be flexibility for specialists to recommend specific vaccines depending on individual risks and blood/serum markers. Live-attenuated vaccines, however, were generally contraindicated [61].
As mentioned, all 50 states now include SCID in neonatal blood tests. For babies diagnosed with SCID, stem cell transplantation has been a potentially curative option. Stem cell transplantation was also described as an option for non-SCID primary immunodeficiencies, including CVID, if patients fail to thrive or develop serious comorbidities such as lymphoma. The low probability of sourcing matched stem cells, potential host rejection, and high mortality rate with the procedure were reported as significant constraints to greater stem cell transplant utilization [65]. However, several clinical trials are currently underway aiming to improve safety and broaden applicability of stem cell transplantation for primary immunodeficiencies, including CVID [66]. Gene therapy has emerged as a potential option for some forms of severe immunodeficiencies, particularly when defects are monogenic [67]. That CVID has been found to be driven by combinations of several genetic (and other) factors makes gene therapy for CVID more complicated and unlikely in the near term.
Cost, Benefit, and Financing of CVID and Treatments
Strong associations were found between the initiation of optimal treatment (including appropriate doses of IgRT and antibiotics) and reduced morbidity and mortality. Financing of treatments, however, was reported as a significant impediment to optimal therapy. Complaints by both patients and physicians were found to be pervasive with regard to health insurer-erected obstacles to coverage, including pre-authorization requirements for every infusion, frequent switching to lowest cost products, and dose restrictions contradicted by evidence [68]. Side effects can be product specific, so switching may negatively affect patients. The Immune Deficiency Foundation received 1417 responses (63% diagnosed with CVID) to its 2014 patient survey on health insurance. Of those, 1160 had been prescribed IgRT for at least 6 months, but 372 reported skipping or delaying at least one dose (82% an average of 2.6 doses) that year. Fifty-two percent of those who skipped or delayed did so because of insurance-related problems [69].
No comprehensive studies were found that specifically assessed CVID-related medical costs before or/and after diagnosis and treatment. A few studies evaluated various categories of infection rates and costs relating to groups of PIDDs. These studies are largely not comparable due to differing cohort selection criteria, timeframes, and basis for averaging (Table 5). Future comprehensive analyses of pre and post-treatment infection and non-infection comorbidity rates and costs by subtype could aid the understanding of treatment intensity and expected benefits at a more granular level; however, the fast-paced discovery of new subtypes of PIDD (described above) would likely complicate comparability of studies over time.
One study, discussed above, compiled PIDD-related hospital expenditures from The Kids’ Inpatient Database, the largest pediatric database in the USA. For the period 2001 to 2012, hospital admissions among B cell subtypes accounted for $342,000,000, averaging $25,357 per admission (Table 3), lower than the average cost of $30,987 across all PIDDs [14]. CVID accounted for 17% of B cell subtype hospitalizations, fewer than the proportion of CVID patients in the Jeffrey Modell Centers Network (27%) (Fig. 1). This disparity does not necessarily imply CVID required fewer admissions than other B cell subtypes. Other potential explanations are that a CVID diagnosis is not recommended before the age of 4, CVID is more likely to be undiagnosed during early childhood, can initiate as other B cell deficiencies and progress into CVID at a later age, and that diagnostic granularity has improved since 2001. Support for these interpretations come from the larger percentage of unspecified hypogammaglobulinemia in the Kids’ database than in the Jeffrey Modell Centers Network, and by very similar totals of CVID, unspecified hypogammaglobulinemia, hyper-IgM syndrome, and selective IgA deficiency in both studies. This study did not distinguish between patients receiving or not receiving optimal therapy prior to hospitalization, so no treatment cost-benefit analysis was possible.
The European quality of life survey mentioned above evaluated CVID cases within a large PIDD cohort and found 343 patients who spent an average of 13.1 days in hospital over 12 months. Of these, the 313 receiving IgRT spent an average of 12.5 days in hospital versus 19.8 (58% more) for the 30 patients not on IgRT [57].
One study of PIDD patients receiving IgRT within a very large administrative database (including privately insured and Medicare covered lives) provided helpful context for analyses of CVID patients on IgRT. In this study, 1076 patients were identified as both diagnosed with PIDD and receiving IgRT for at least 6 months. Of these, nearly half (48%) had a diagnosis of CVID, evidencing the large proportion of CVID within the IgRT-receiving PIDD population as a whole. This entire cohort had a per-patient-year infection rate (pneumonia and bronchitis) of 37% despite stable IgRT. Infection rates prior to initiation of IgRT were not reported, so no pre-post comparisons could be made [70]. Another study of a large medical claims database identified 490 patients both diagnosed with PIDD and receiving IgRT, and who had suffered from at least one infection over a 7-month period. Infection-related medical expenditures (including hospitalizations) for these 490 patients totaled over $10,000,000, an annualized average of $20,443. The average cost of a hospital stay was $27,952 [71]. Costs prior to IgRT initiation were not reported so no pre-post-treatment analysis was possible.
One analysis was found that compared pre and post-diagnosis medical costs for a large group of PIDD patients treated within the Jeffrey Modell Centers Network. SCID was excluded, the intensive treatment requirements for which would have skewed average costs. A reasonable estimate of percentage of CVID cases included in the study, while not disclosed, would be 18%, greater than the Jeffrey Modell Centers Network’s 15.4% due to exclusion of SCID (Fig. 1). The authors calculated average pre-diagnosis annual cost as $111,053, which included treatment for infections, physician/emergency room visits, hospital days, and work or school days missed. Post-diagnosis annual cost averaged $25,171, before inclusion of IgRT. IgRT was estimated as an additional $30,000 for a total cost of $55,171. Net average annual per-patient savings including and excluding IgRT, achievable by diagnosing and treating PIDD, was $85,882 and $55,882, respectively (Table 4) [13••]. This high level of savings from treatment could support aggressive searches by payors within their databases for undiagnosed PIDD populations, and could justify the cost of incremental screenings for identified, at-risk cases. The report did not include material supporting data or methodologies used; additional peer-reviewed studies quantifying health system savings from treatment could further incentivize payors to find and treat cases of PIDD within their membership pools (Table 5).
Conclusions
Heterogeneity and overlap in clinical manifestations within, between, and unrelated to immune disorders were found to confound timely symptom recognition of CVID. Therefore, a reliable, widely accepted framework for early CVID identification remains elusive. A logical objective of clinicians should be to diagnose early to initiate treatment before chronic, recurrent, or severe infections cause irreversible organ damage. Advancements in diagnostic technology and consensus guidelines have allowed classification of primary immunodeficiencies, including CVID, to become more robust. While only a small percentage of CVID cases have identifiable genetic drivers currently, laboratory tests that identify immunoglobulin deficiencies are widely available, informing requisite initiation of the most important treatment available for CVID, namely, IgRT. Prophylactic and aggressive antibiotic therapies may be indicated for persistent or breakthrough infections once patients are known to have CVID. Today it is understood that patients should be monitored and assessed regularly by their PCPs and specialists for potential protocol adjustments to optimize long-term health. Risk stratification may help guide these decisions. Available treatments and protocols have made improved health outcomes and quality of life possible for those with CVID.
IgRT product enhancements and administration options have improved significantly over time, providing greater convenience and treatment compliance. IgRT protocols have moved to higher or “biologic doses” and are individualized to suppress rates of infections. The evidence of IgRT effectiveness in CVID is very strong, but it is rarely functionally curative. Those with CVID continue to have certain types of infections—especially of the upper respiratory and sinuses—that require medical attention. This, together with the high cost of IgRT, may be contributing to insurance coverage barriers encountered by many patients and PCPs. Peer-reviewed studies are needed to support the compelling cost savings achieved through treatment. If savings are substantiated, insurers may be financially incentivized to proactively review their insured databases for potentially undiagnosed CVID and other PIDD cases.
The nature of the CVID symptom recognition is likely to evolve. As the cost of next generation sequencing falls, CVID registries grow in size and breadth, and bioinformatics capabilities uncover multifactorial drivers of CVIDs, PCPs may someday have periodic, routine laboratory tests to guide them. In the meantime, however, symptom recognition by PCPs will remain a gating factor for necessary treatments. Accepted, effective symptom recognition frameworks, along with sufficient PCP education and training, are needed to optimize health for those with CVID.
Abbreviations
- Ig:
-
Immunoglobulin
- IgRT:
-
Immunoglobulin replacement therapy
- PCP:
-
Primary care practitioner (here, including emergency room practitioners)
- PIDD:
-
Primary immunodeficiency
- SCID:
-
Severe combined immunodeficiency
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Ochs HD, Hitzig WH. History of primary immunodeficiency diseases. Curr Opin Allergy Clin Immunol. 2012;12(6):577–87.
Boyle JM, Buckley RH. Population prevalence of diagnosed primary immunodeficiency diseases in the United States. J Clin Immunol. 2007;27:497–502.
Fudenberg H, Good RA, Goodman HC, Hitzig W, Kunkel HG, Roitt IM, et al. Primary immunodeficiencies, report of a World Health Organization committee. Pediatrics. 1971;47(5):927–46.
• Bonilla F, Barlan I, Chapel H, Costa-Carvalho B, Cunningham-Rundles C, de La Morena M, et al. International consensus document (ICON): common variable immunodeficiency disorders. J Allergy Clin Immunol Pract. 2016;4(1):38–59 A comprehensive overview of CVID, including epidemiology, clinical manifestations, genetics, diagnosis, treatments and prognosis, resulting from extensive international collaboration.
Bousfiha AA, Jeddane L, Picard C, Ailal F, Gaspar HB, Al-Herz W, et al. The 2017 IUIS phenotypic classification for primary immunodeficiencies. J Clin Immunol. 2018;38:128–43.
Tangye SG.. Inborn errors of immunity committee (IEI)—activities; 2018. Retrieved February, 2019, from http://www.iuisonline.org/index.php?option=com_content&view=article&id=68&Itemid=94&5a48d87dcdf6fac96a6bc0ffc1b9e64c=461e23649300226f90ec7f639f96ce00.
Immune Deficiency Foundation, prepared by Schulman, Ronca & Bucuvalas, Inc. Primary immune deficiency diseases in America, the first national survey of patients and specialists, No. IDF0034, Towson, Maryland. 1995.
Joshi AY, Iyer VN, Hagan JB, St. Sauver JL, Boyce TG. Incidence and temporal trends of primary immunodeficiency: a population-based cohort study. Mayo Clin Proc. 2009;84(1):16–22.
Kobrynski L, Powell RW, Bowen S. Prevalence and morbidity of primary immunodeficiency diseases, United States 2001–2007. J Clin Immunol. 2014;34(8):954–61.
National Institutes of Health (NIH). National Institutes of Health (NIH), National Institute of Allergy and Infectious Diseases—primary immune deficiency diseases (PIDDs); 2016. Retrieved February 10, 2019, from https://www.niaid.nih.gov/diseases-conditions/primary-immune-deficiency-diseases-pidds.
Bousfiha AA, Jeddane L, Ailal F, Benhsaien I, Mahlaoui N, Casanova JL, et al. Primary immunodeficiency diseases worldwide: more common than generally thought. J Clin Immunol. 2013;33:1–7.
Jeffrey Modell Foundation (JMF). Our story. Retrieved 01/15, 2019, from http://www.info4pi.org/hq/our-story
•• Modell V, Quinn J, Orange J, Notarangelo LD, Modell F. Primary immunodeficiencies worldwide: an updated overview from the Jeffrey Modell Centers global network. Immunol Res. 2016;64:736–53 A unique analysis showing PIDD-related costs for a large patient population prior to and post diagnosis.
Rubin Z, Pappalardo A, Schwartz A, Antoon JW. Prevalence and outcomes of primary immunodeficiency in hospitalized children in the United States. J Allergy Clin Immunol in Pract. 2018;6(5):1705–10.
Cunningham-Rundles C, Sidi P, Estrella L, Doucette J. Identifying undiagnosed primary immunodeficiency diseases in minority subjects by using computer sorting of diagnosis codes. J Clin Immunol. 2004;113:747–55.
Immune Deficiency Foundation (IDF). All 50 states now screening newborns for severe combined immunodeficiency (SCID); 2018. Retrieved 02/04/2019, 2019, from https://www.primaryimmune.org/news/all-50-states-now-screening-newborns-severe-combined-immunodeficiency-scid.
Griffith LM, Cowan MJ, Notarangelo LD, Kohn DB, Puck JM, Shearer WT, et al. Primary immune deficiency treatment consortium (PIDTC) update. J Clin Immunol. 2016;138(2):375–85.
Chapel H, Lucas M, Patel S, Lee M, Cunningham-Rundles C, Resnick E, et al. Confirmation and improvement of criteria for clinical phenotyping in common variable immunodeficiency disorders in replicate cohorts. J Clin Immunol. 2012;130:1197–8.
• Modell V, Orange JS, Quinn J, Modell F. Global report on primary immunodeficiencies: 2018 update from the Jeffrey Modell Centers Network on disease classification, regional trends, treatment modalities, and physician reported outcomes. Immunol Res. 2018;66:367–80 A comprehensive breakdown of PIDD subtypes within a large patient population.
Waltenburg R, Kobrynski L, Reyes M, Bowen S, Khoury MJ. Primary immunodeficiency diseases: practice among primary care providers and awareness among the general public, United States, 2008. Genet Med. 2010;12(12):792–800.
Verbsky JW, Routes JR. Recurrent fever, infections, immune disorders, and autoinflammatory diseases. In: Kliegman RM, Lye PS, Bordini BJ, Toth H, Basel D, editors. Nelson pediatric symptom-based diagnosis. Amsterdam: Elsevier; 2018. p. 746–73.
Abolhassani H, Sagvand B, Shokuhfar T, Mirminachi B, Rezaei N, Aghamohammadi A. A review on guidelines for management and treatment of common variable immunodeficiency. Expert Rev Clin Immunol. 2013;9(6):561–75.
Ameratunga R, Woon ST, Gillis D, Koopmans W, Steele R. New diagnostic criteria for common variable immune deficiency (CVID), which may assist with decisions to treat with intravenous or subcutaneous immunoglobulin. Clin Exp Immunol. 2013;174:203–11.
Urschel S, Kayikci L, Wintergerst U, Notheis G, Jansson A, Belohradsky B. Common variable immunodeficiency disorders in children: delayed diagnosis despite typical clinical presentation. J Pediatr. 2009;154(6):888–94.
Cunningham-Rundles C. How I treat common variable immune deficiency. Blood. 2010;116(1):7–15.
Chapel H. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood. 2008;112(2):277–86.
Grundling K. Common variable immunodeficiency in the very old. J Allergy Clin Immunol. 2014;133(2):AB9.
Cunningham-Rundles C. The many faces of common variable immunodeficiency, vol. 2012: Hematology Am Soc Hematol Educ Program; 2012. p. 301–5.
Kardar GA, Oraei M, Shahsavani M, Namdar Z, Kazemisefat GE, Haghi Ashtiani MT, et al. Reference intervals for serum immunoglobulins IgG, IgA, IgM and complements C3 and C4 in Iranian healthy children. Iran J Public Health. 2012;41(7):59–63.
Welch K, Resnick E, Cunningham-Rundles C. Genetic basis of CVID-like disease. J Allergy Clin Immunol. 2014;133(2) supplement.
Schouwenburg PA, Davenport EE, Kienzler AK, Marwah I, Wright B, Lucas M, et al. Application of whole genome and RNA sequencing to investigate the genomic landscape of common variable immunodeficiency disorders. Clin Immunol. 2015;160:301–14.
de Valles-Ibáñez G, Esteve-Solé A, Piquer M, González-Navarro EA, Hernandez-Rodriguez J, Laayouni H, et al. Evaluating the genetics of common variable immunodeficiency: monogenetic model and beyond. Front Immunol. 2018;9(636):1–15.
Li J, Wei Z, Li YR, Maggadottir SM, Chang X, Desai A, et al. Understanding the genetic and epigenetic basis of common variable immunodeficiency disorder through omics approaches. Biochim Biophys Acta. 2016;1860:2656–63.
Westh L, Mogensen TH, Dalgaard LS, Bernth Jensen JM, Katzenstein T, Hansen ABE, et al. Identification and characterization of a nationwide Danish adult common variable immunodeficiency cohort. Scand J Immunol. 2017;85:450–61.
Orange JS, Grossman W, Navickis R, Wilkes M. Impact of trough IgG on pneumonia incidence in primary immunodeficiency: a meta-analysis of clinical studies. Clin Immunol. 2010;137(1):21–30.
Cinetto F, Scarpa R, Rattazzi M, Agostini C. The broad spectrum of lung diseases in primary antibody deficiencies. Eur Respir Rev. 2018;27(149):180019.
• Odnoletkova I, Kindle G, Quinti I, Grimbacher B, Knerr V, Gathmann B, et al. The burden of common variable immunodeficiency disorders: a retrospective analysis of the European society for immunodeficiency (ESID) registry data. Orphanet J Rare Dis. 2018;13(201):1–17 A large and unique study analyzing individual and society disease burden of CVID.
Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood. 2011;119(7):1650–7.
Quinti, I., Soresina A, Spadaro G, Martino S, Donnanno S, Agostini C, et al. (2007). Long-term follow-up and outcome of a large cohort of patients with common variable immunodeficiency. J Clin Immunol, 27(3), 308–316.
Blaese R, Stiehm E, Bonilla F, Younger M, editors. Immune Deficiency Foundation patient & family handbook for primary immunodeficiency diseases. 5th ed. Baltimore: Immune Deficiency Foundation; 2013.
Maarschalk-Ellerbroek LJ, Hoepelman AIM, van Montfrans JM, Ellerbroek PM. The spectrum of disease manifestations in patients with common variable immunodeficiency disorders and partial antibody deficiency in a university hospital. J Clin Immunol. 2012;32:907–21.
Gathmann, B., & Mahlaoui N., , Gérard L., Oksenhendler E., Warnatz K., Schulze I., Kindle G., Kuijpers T.W., van Beem R., Guzman D., Workman S., Soler-Palacín P., de Gracia J., Witte T., Schmidt R.E., Litzman J., Hlavackova E., Thon V., Borte M., Borte S., Kumararatne D., Feighery C., Longhurst H., Helbert M., Szaflarska A., Sediva A., Belohradsky B.H., Jones A., Baumann U., Meyts I., Kutukculer N., Wågström P., Galal N.M., Roesler J., Farmaki E., Zinovieva N., Ciznar P., Papadopoulou-Alataki E., Bienemann K., Velbri S., Panahloo Z., Grimbacher B., CEREDIH., Dutch WID, European Society for Immunodeficiencies Registry Working Party (2014). Clinical picture and treatment of 2212 patients with common variable immunodeficiency. J Allergy Clin Immunol, 134(1), 116–126.
Modell V, Quinn J, Ginsberg G, Gladue R, Orange J, Modell F. Modeling strategy to identify patients with primary immunodeficiency utilizing risk management and outcome measurement. Immunol Res. 2017;65:713–20.
Bjelac JA, Yonkof JR, Fernandez J. Differing performance of the warning signs for immunodeficiency in the diagnosis of pediatric versus adult patients in a two-center tertiary referral population. J Clin Immunol. 2018.
Arkwright PD, Gennery AR. Ten warning signs of primary immunodeficiency: a new paradigm is needed for the 21st century. Ann N Y Acad Sci. 2011;1238:7–14.
Subbarayan A, Colarusso G, Hughes SM, Gennery AR, Slatter M, Cant AJ, et al. Clinical features that identify children with primary immunodeficiency diseases. Pediatrics. 2011;127(5):810–6.
Lankisch P, Schiffner J, Ghosh S, Babor F, Borkhardt A, Laws HJ. The Dusseldorf warning signs for primary immunodeficiency: is it time to change the rules? J Clin Immunol. 2015;35:273–9.
de Vries E, et al. Patient-centred screening for primary immunodeficiency, a multi-stage diagnostic protocol designed for non-immunologists: 2011 update. Clin Exp Immunol. 2011(167):108–19.
Orange JS, Seeborg FO, Boyle M, Scalchunes C, Hernandez-Trujillo V. Family physician perspectives on primary immunodeficiency diseases. Front Med. 2016;3(12).
Hernandez-Trujillo VP, Scalchunes C, Hernandez-Trujillo HS, Boyle J, Williams P, Boyle M, et al. Primary immunodeficiency diseases: an opportunity in pediatrics for improving patient outcomes. Clin Pediatr. 2015;54(13):12652015–1275.
American Academy of Allergy, Asthma, & Immunology (AAAAI); 2019. Retrieved 02/15, 2019, from https://education.aaaai.org/immunodeficiency/2018DiagnosticSchool#group-tabs-node-course-default1.
Reust CE. Evaluation of primary immunodeficiency disease in children. Am Fam Physician. 2013;87(11):773–8.
Orange JS, Glessner JT, Resnick E, Sullivan KE, Lucas M, Ferry B, et al. Genome-wide association identifies diverse causes of common variable immunodeficiency. J Allergy Clin Immunol. 2011;127(6):1360–7.
Perez EE, Orange JS, Bonilla F, Chinen J, Chinn IK, Dorsey M, et al. Update on the use of immunoglobulin in human disease: a review of evidence. J Allergy Clin Immunol. 2017;139:S1–S46.
Berger M. Principles of and advances in immunoglobulin replacement therapy for primary immunodeficiency. Immunol Allergy Clin N Am. 2008;28:413–37.
Pulvirenti F, Cinetto F, Pecoraro A, Carrabba M, Crescenzi L, Neri R, et al. Health-related quality of life in patients with CVID under different schedules of immunoglobulin administration: prospective multicenter study. J Clin Immunol. 2019;39:159–70.
Eades-Perner AM, Gathmann B, Knerr V, Guzman D, Veit D, Kindle G, et al. The European internet-based patient and research database for primary immunodeficiencies: results 2004–06. Clin Exp Immunol. 2007;147:306–12.
Bonagura VR, Marchlewski R, Cox A, Rosenthal DW. Biologic IgG level in primary immunodeficiency disease: the IgG level that protects against recurrent infection. J Clin Immunol. 2008;122:210–2.
Lucas M, Lee M, Lortan J, Lopez-Granados E, Misbah M, Chapel H. Infection outcomes in patients with common variable immunodeficiency disorders: relationship to immunoglobulin therapy over 22 years. J Allergy Clin Immunol. 2010;125(6):1354–60.
Langereis JD, van der Flier M, de Jonge MI. Limited innovations after more than 65 years of immunoglobulin replacement therapy: potential of IgA- and IgM-enriched formulations to prevent bacterial respiratory tract infections. Front Immunol. 2018;9(1925):1–8.
Schroeder HW Jr, Cavacini L. Structure and function of immunoglobulins. J Allergy Clin Immunol. 2010;125(202):S41–52.
Quinti I, Soresina A, Guerra A, Rondelli R, Spadaro G, Agostini C, et al. Effectiveness of immunoglobulin replacement therapy on clinical outcome in patients with primary antibody deficiencies: results from a multicenter prospective cohort study. J Clin Immunol. 2011;31:315–22.
Kuruvilla M, de la Morena MT. Antibiotic prophylaxis in primary immune deficiency disorders. J Allergy Clin Immunol Pract. 2013;1:573–82.
Sperlich JM, Grimbacher B, Workman S, Haque T, Seneviratne SL, Burns S, et al. Respiratory infections and antibiotic usage in common variable immunodeficiency. J Allergy Clin Immunol. 2018;6:159–68.
Wehr, C., Gennery AR, Lindemans C, Schulz A, Hoenig M, Marks R, et al. (2014). Multicenter experience in hematopoietic stem cell transplantation for serious complications of common variable immunodeficiency. J Allergy Clin Immunol, 135(4), 988–997.
Clinicaltrials.gov; 2019. Stem cell transplantation, retrieved 02/11/2019, from https://clinicaltrials.gov/ct2/results?pg=1&load=cart&id=NCT02349906+OR+NCT02990819+OR+NCT01966367+OR+NCT02061800+OR+NCT00919503+OR+NCT02065869+OR+NCT01962415+OR+NCT03301168.
Clinicaltrials.gov. 2019, gene therapy trials for primary immunodeficiencies, retrieved 02/04/2019, from https://clinicaltrials.gov/ct2/results?pg=1&load=cart&id=NCT03538899+OR+NCT01306019+OR+NCT03601286+OR+NCT03315078+OR+NCT01512888+OR+NCT03645460+OR+NCT03311503+OR+NCT03217617+OR+NCT01347242+OR+NCT01906541+OR+NCT02234934+OR+NCT02757911+OR+NCT01855685+OR+NCT03812263+OR+NCT02333760+OR+NCT03645486.
Shapiro RS, Boyle M. Payor issues: barriers to optimal management of patients with primary immunodeficiency. J Clin Immunol. 2012;32(Supplement 2):S410–4.
Lyles CA, Scalchunes C, Boyle M, Henderson T, LaMotte L. Health insurance & primary immunodeficiency diseases: a 2014 Immune Deficiency Foundation survey (Survey. Towson, Maryland: Immune Deficiency Foundation. (Primary immunodeficiency health insurance survey). 2014.
Wasserman RL, Ito D, Xiong V, Ye XL, Bonnet P, Li-McLeod J. Impact of site of care on infection rates among patients with primary immunodeficiency diseases receiving intravenous immunoglobulin therapy. J Clin Immunol. 2017;37:180–6.
Menzin J, Sussman M, Munsell M, Zbrozek A. Economic impact of infections among patients with primary immunodeficiency disease receiving IVIG therapy. Clinicoecon Outcomes Res. 2014;6:297–302.
Further Reading
Baris S, Ercan H, Cagan H, Ozen A, Karakoc-Aydiner E, Ozdemir C, et al. Efficacy of intravenous immunoglobulin treatment in children with common variable immunodeficiency. J Investig Allergol Clin Immunol. 2011;21(7):514–21.
Bonilla FA, Khan DA, Ballas ZK, Chinen JC, Frank MM, Hsu JT, et al. Practice parameter for the diagnosis and management of primary immunodeficiency. J Allergy Clin Immunol. 2015;136(5):1186–205.
Buckley RH, editor. Immune Deficiency Foundation diagnostic & clinical care guidelines for primary immunodeficiency diseases. 3rd ed. Towson: Immune Deficiency Foundation; 2015.
Busse P, Razvi S, Cunningham-Rundles C. Efficacy of intravenous immunoglobulin in the prevention of pneumonia in patients with common variable immunodeficiency. J Allergy Clin Immunol. 2002;109(6):1001–4.
Chapel H, Cunningham-Rundles C. Update in understanding common variable immunodeficiency disorders (CVIDs) and the management of patients with these conditions. Br J Haematol. 2009;145:709–29.
Llobet M, Soler-Palacin P, Detkova D, Hernández M, Caragol I, Espanol T. Common variable immunodeficiency: 20-yr experience at a single centre. Pediatr Allergy Immunol. 2009;20(2):113–8.
Modell F, Puente D, Modell V. From genotype to phenotype. Further studies measuring the impact of a physician education and public awareness campaign on early diagnosis and management of primary immunodeficiencies. Immunol Res. 2009;38:132–49.
Orange J, Ballow M, Stiehm R, Ballas Z, Chinen J, De La Morena M, et al. Use and interpretation of diagnostic vaccination in primary immunodeficiency: a working group report of the basic and clinical immunology interest section of the American Academy of Allergy, Asthma & Immunology. The J Allergy Clin Immunol. 2012;130(3):S1–S24.
Park, C. (2018). Pediatric common variable immunodeficiency. Retrieved October 5, 2018, from https://emedicine.medscape.com/article/885935-overview
Samples S, Sharma N, Ownby D. New diagnosis of common variable immunodeficiency in a 12-year-old with pneumonia: an illustrative case. Hospital Pediatrics. 2014;4(4):251–5.
Shapiro RS, Wasserman R, Bonagura V, Gupta S. Emerging paradigm of primary immunodeficiency disease: individualizing immunoglobulin dose and delivery to enhance outcomes. J Clin Immunol. 2017;37:190–6.
Shehata N, Palda V, Bowen T, Haddad E, Issekutz T, Mazer B, et al. The use of immunoglobulin therapy for patients with primary immune deficiency: an evidence-based practice guideline. Transfus Med Rev. 2010;24(1):S28–50.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
William Gerber declares no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Critical Care
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Gerber, W.A. Focus on Chronic Variable Immunodeficiency for Primary Care Practitioners, the Gatekeepers to Optimal Health Outcomes for Primary Immunodeficiency Syndromes. Curr Pediatr Rep 7, 130–144 (2019). https://doi.org/10.1007/s40124-019-00202-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40124-019-00202-8