
Abstract
Introduction
Vaccination is a critical tool for preventing coronavirus disease 2019 (COVID-19) and influenza illnesses. Coadministration of the COVID-19 vaccine, BNT162b2, with seasonal inactivated influenza vaccine (SIIV) can provide substantial benefits, including streamlining vaccine delivery.
Methods
In this phase 3 study, healthy 18- to 64-year-olds who had received three previous doses of BNT162b2 were randomized (1:1) to the coadministration group (month 0, BNT162b2 + SIIV; month 1, placebo) or the separate-administration group (month 0, placebo + SIIV; month 1, BNT162b2). The primary immunogenicity objective was to demonstrate that the immune responses elicited by BNT162b2 and SIIV [measured by full-length S-binding immunoglobulin G (IgG) levels and strain-specific hemagglutination inhibition assay (HAI) titers against four influenza strains 1 month post-vaccination, respectively] when coadministered were noninferior to those elicited by either vaccine administered alone, based on a prespecified 1.5-fold noninferiority margin [lower bound 95% CI for geometric mean ratio (GMR) > 0.67]. Reactogenicity and adverse event (AE) rates were evaluated.
Results
Randomized participants who received study vaccination (N = 1128; coadministration group, n = 564; separate-administration group, n = 564) had a median age of 39 years. Model-adjusted GMRs for coadministration to separate administration were 0.83 (95% CI 0.77, 0.89) for full-length S-binding IgG levels and 0.89–1.00 (lower bound of all 95% CIs > 0.67) for the four influenza strain-specific HAI titers, with all endpoints achieving the prespecified noninferiority criterion. Reactogenicity events were mostly mild or moderate when BNT162b2 was coadministered with SIIV. Serious AEs were reported in < 1% of participants within 1 month after any vaccination; none were considered vaccine-related.
Conclusions
BNT162b2 coadministered with SIIV elicited immune responses that were noninferior to those elicited by BNT162b2 alone and SIIV alone, and BNT162b2 had an acceptable safety profile when coadministered with SIIV. The results of this study support the coadministration of BNT162b2 and SIIV in adults.
Trial Registration
ClinicalTrials.gov registration: NCT05310084.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Why carry out this study? |
Vaccination is a critical tool for preventing COVID-19 and influenza illnesses. |
Coadministration of the BNT162b2 COVID-19 vaccine with seasonal inactivated influenza vaccine (SIIV) can streamline vaccine delivery. |
To support the coadministration of BNT162b2 and SIIV in adult populations, this study investigated the safety, tolerability, and immunogenicity of a fourth dose (booster) of BNT162b2 administered concomitantly with SIIV or administered separately 1 month after SIIV in adults who had received three previous doses of BNT162b2. |
BNT162b2 coadministered with SIIV elicited noninferior immune responses compared with those elicited by BNT162b2 alone and SIIV alone, and BNT162b2 had an acceptable safety profile when coadministered with SIIV. |
What has been learned from the study? |
The results of this study support the coadministration of BNT162b2 and SIIV in adults; provision of rigorous clinical trial data should help contribute to the acceptance of influenza vaccine and COVID-19 vaccine coadministration. |
Introduction
Widespread use of safe and effective vaccines has been critical to curtailing the COVID-19 pandemic. The efficacy and safety of two doses of the BNT162b2 mRNA COVID-19 vaccine was established in a global phase 2/3 trial in those 16 years and older, with vaccine efficacy of 95.0% [1]. In a phase 3 study, the efficacy of a third BNT162b2 dose given ≥ 6 months after the last previous dose was 95.3% among those 16 years and older, and reduced rates of SARS-CoV-2 infection and severe COVID-19 among older adults who had received their last dose at least 5 months prior [2, 3]. Effectiveness of COVID-19 vaccine booster doses against milder disease wanes over months, but remains high against severe disease and mortality when the vaccine is more closely matched to circulating SARS-CoV-2 variants [4,5,6,7]. Data also indicate that COVID-19 displays seasonal epidemiologic patterns, predominating in the winter season [8, 9]. Many countries currently recommend seasonal COVID-19 vaccine boosters to address future disease waves and the emergence of highly transmissible and potentially virulent strains [10,11,12,13,14].
Influenza is another seasonal respiratory disease that causes yearly epidemics, and occasionally pandemics, and is associated with substantial morbidity and mortality, particularly among older adults, very young children, those who are pregnant, and those with certain chronic medical conditions [15, 16]. In temperate regions, influenza transmission occurs predominantly in the winter season.
As the COVID-19 pandemic abates and seasonality of viral respiratory disease returns to more typical patterns, there is a risk that peaks of influenza, COVID-19, and respiratory syncytial virus illnesses will occur, particularly in winter, straining healthcare resources [17]. Although with an estimated prevalence of < 1% [18], coinfection with SARS-CoV-2 and the influenza virus may also occur [19], which could lead to adverse outcomes [17, 19,20,21]. Together, these prospects reinforce the importance of vaccination against both COVID-19 and influenza.
Seasonal vaccination against influenza represents the primary strategy for preventing influenza illness and its complications [16]. Effectiveness of influenza vaccination is dependent on several factors, including the similarity between circulating influenza viruses and the virus strains covered by the seasonal vaccine. Accordingly, annual influenza vaccine updates are necessitated by the typical peak of illness in winter months and the constant evolution of the influenza virus, to support vaccines more closely matched to circulating strains [22]. Adult annual seasonal influenza vaccination is recommended in many countries, particularly for individuals with risk factors for increased influenza morbidity and mortality, such as older adults and those with chronic medical conditions [16, 23, 24].
Because recommendations for COVID-19 vaccination and influenza vaccination have considerable overlap, including recommended age groups, at-risk populations, and timing, both vaccines may need to be administered at the same time. Such guidance is already provided in some jurisdictions, including the USA and European Union, and by the World Health Organization (WHO) [10, 24,25,26,27]. This is based on the principle that two inactivated vaccines can be given concurrently and with the aim of maximizing the opportunity to receive both vaccines, and eliminating the need for more than one healthcare visit for those receiving both vaccines, which will be more convenient for the healthcare provider and the vaccine recipient and may lead to improved vaccination rates.
To investigate the coadministration of BNT162b2 and seasonal inactivated influenza vaccine (SIIV) in adult populations, this study assessed the safety, tolerability, and immunogenicity of a fourth dose (booster) of BNT162b2 administered either concomitantly with SIIV or separately 1 month after SIIV in adults who had received three previous doses of BNT162b2.
Methods
Study Design and Participants
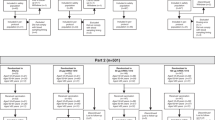
This was a phase 3, multicenter, randomized, observer-blind study conducted at 25 sites in Australia and New Zealand from 20 April to 5 October 2022 (NCT05310084), and was initiated before the introduction of Omicron-adapted vaccines. Using interactive response technology, participants were randomized 1:1 to either the coadministration group (BNT162b2 + SIIV administered at month 0 and placebo administered at month 1) or the separate-administration group (placebo + SIIV administered at month 0 and BNT162b2 administered at month 1; Fig. 1). Randomized participants were stratified by age group (18−49 years and 50−64 years) and by history (versus no history) of a positive SARS-CoV-2 test result (by either nucleic acid amplification test or rapid antigen test) before randomization.

Study design. SIIV seasonal inactivated influenza vaccine
Healthy participants who were 18–64 years of age, including those with any preexisting stable disease (i.e., not requiring significant therapy modification or hospitalization for worsening disease during the 6 weeks and 12 weeks before enrollment, respectively), were eligible to participate. Participants had received three previous doses of 30 µg BNT162b2, with the third dose administered ≥ 90 days before study vaccination. Participants and their partners were to use appropriate contraception during the intervention period and for 28 days after the last vaccination. Key exclusion criteria included immunocompromise with known or suspected immunodeficiency as determined by history and/or laboratory or physical examination, receipt of radiotherapy or immunosuppressive treatment, a history of Guillain–Barré syndrome or of severe allergic reaction to a vaccine, a positive SARS-CoV-2 test result within 28 days before the first vaccination, being pregnant or breastfeeding, receipt of influenza vaccine within 6 months before study vaccination, receipt of any COVID-19 vaccine other than BNT162b2, or receipt of more than three previous BNT162b2 doses (see Supplementary Material for additional exclusion criteria, vaccination temporary delay criteria, and prohibited vaccines and medications during the study).
Ethical Approval
The study was conducted in accordance with the consensus ethical principles derived from international guidelines, including the Declaration of Helsinki and Council for International Organizations of Medical Sciences International Ethical Guidelines, as well as applicable International Council for Harmonisation Good Clinical Practice guidelines, laws, and regulations, including privacy laws. The ethics committees for this study were Monash Health Human Research Ethics Committee (Australia; Monash Health Local Reference RES-22-0000-042A), which approved the study and protocol amendments, and the COVID-19 Emergency Standard Operating Procedures of the New Zealand Health and Disability Committee (New Zealand; Ethics Reference 2021 FULL 11293), which approved the study, and the Northern B Health and Disability Ethics Committee (New Zealand; Ethics Reference 2022 AM 11293), which approved subsequent protocol amendments. All participants who received study vaccinations provided written informed consent.
Intervention and Blinding
Commercially available, licensed quadrivalent SIIV was used [Afluria Quad; Seqirus Pty Ltd, Victoria, Australia, and Seqirus (NZ) Ltd, Auckland, New Zealand]. The SIIV for the 2022 season contained 15 μg hemagglutinin of each of the following: A/Victoria/2570/2019 (H1N1)pdm09-like virus; A/Darwin/9/2021 (H3N2)-like virus; B/Austria/1359417/2021-like virus; and B/Phuket/3073/2013-like virus. Original BNT162b2 30 µg, placebo (0.9% NaCl), and SIIV were administered intramuscularly. For the first vaccination, BNT162b2 (coadministration group) or placebo (separate-administration group) was administered to the left deltoid, and SIIV (both groups) was administered to the right deltoid (Fig. 1). For the second vaccination, placebo (coadministration group) or BNT162b2 (separate-administration group) was administered to the left deltoid.
In this observer-blinded study, staff receiving, storing, dispensing, preparing, and administering the study vaccines were unblinded. All other study and site personnel, including the investigator and investigator staff, as well as participants, were blinded to study vaccine assignments (i.e., BNT162b2 or placebo). In particular, the individuals who evaluated participant safety were blinded. Because of differences in their physical appearance, the study vaccines were administered in a manner that prevented participants from identifying the study vaccine group on the basis of its appearance.
Immunogenicity Objectives and Assessments
To assess immunogenicity, approximately 30 mL of blood was collected immediately before the first vaccination and then immediately before and 1 month after the second vaccination. The primary immunogenicity objective was to demonstrate that immune responses elicited by BNT162b2 when coadministered with SIIV were noninferior to those elicited by either vaccine administered alone. The primary BNT162b2 immunogenicity endpoint was the geometric mean ratio (GMR) of full-length S-binding immunoglobulin G (IgG) levels 1 month after BNT162b2 vaccination in the coadministration group to 1 month after BNT162b2 vaccination in the separate-administration group. The primary SIIV immunogenicity endpoints were the GMRs of strain-specific hemagglutination inhibition assay (HAI) titers 1 month after receiving SIIV in the coadministration group to the corresponding assays 1 month after receiving SIIV in the separate-administration group.
Secondary immunogenicity endpoints included geometric mean concentrations (GMCs) for full-length S-binding IgG levels or geometric mean titers (GMTs) for strain-specific HAI titers before and 1 month after receipt of BNT162b2 or SIIV, respectively, and the geometric mean-fold rise (GMFR) from before to 1 month after receipt of BNT162b2 or SIIV. Exploratory objectives included evaluation of seroprotection and seroconversion rates after SIIV when coadministered with BNT162b2 or when administered with placebo. An additional secondary BNT162b2 immunogenicity endpoint was SARS-CoV-2 neutralizing titers, which will be reported separately.
Safety Objectives and Assessments
The primary safety objective was to evaluate the safety profile of BNT162b2 when coadministered with SIIV. Local reactions (at the BNT162b2 or placebo injection sites only) and systemic events (including fever and antipyretic use) occurring within 7 days after each injection were recorded by participants in an electronic diary. SIIV injection site reactions were collected as adverse events (AEs; not in the electronic diary). Data regarding AEs and serious AEs (SAEs) occurring within 1 month after each vaccination were collected, and investigators assessed if AEs had a causal relationship to study vaccine (BNT162b2 or placebo). Acute reactions (reactogenicity or AEs) occurring within 30 min of vaccination were counted as immediate AEs. AEs were categorized by the Medical Dictionary for Regulatory Activities v25.1 terms.
Statistics
The sample size determination is provided in the Supplementary Material. The study analysis populations are summarized in Table S1.
Immunogenicity endpoints were assessed in the evaluable immunogenicity populations. For the primary endpoints, model-adjusted GMRs and associated 95% CIs were calculated by exponentiating the difference in least squares means of the two vaccine groups and the corresponding CIs based on analysis of logarithmically transformed assay results using a linear regression model that included vaccine group, age group, and corresponding baseline full-length S-binding IgG level or strain-specific HAI titer results as covariates.
For the primary BNT162b2 and SIIV immunogenicity objectives, noninferiority was declared if the lower bound of the two-sided 95% CI for the model-adjusted GMR was > 0.67. The primary SIIV immunogenicity objective was achieved if the noninferiority criterion was met for each of the four assessed influenza strains (i.e., B/Austria, B/Phuket, H1N1 A/Victoria, and H3N2 A/Darwin).
The GMFRs were calculated by exponentiating the mean of the difference of logarithmically transformed assay results (later time point minus earlier time point). The associated two-sided 95% CIs were obtained by constructing CIs using the Student’s t-distribution for the mean difference on the logarithmic scale and exponentiating the CIs.
Geometric means were calculated as the mean of the assay results after making the logarithm transformation and then exponentiating the mean to express results on the original scale. Two-sided 95% CIs were obtained by taking log transformations of assay results, calculating the 95% CI with reference to the Student’s t-distribution, and then exponentiating the CIs.
The primary safety objective was evaluated by descriptive summary statistics after each vaccination and for each vaccine group using the safety population. Descriptive statistics for categorical variables included the percentages and associated Clopper–Pearson 95% CIs.
Results
Participants
The study randomized 1134 participants (coadministration group, n = 568; separate-administration group, n = 566); 560 (98.6%) and 555 (98.1%) participants completed the study, respectively (Fig. 2). Baseline demographics were similar for the coadministration and separate-administration groups (Table 1). Overall, 63.6% (717/1128) of participants were female, 78.5% (885/1128) were white, and the median (range) age at first study vaccination was 39 (18–65) years; 72.2% (814/1128) of participants were 18–49 years of age. The majority of participants had a negative baseline SARS-CoV-2 infection status [65.7% (741/1128)]. The median (range) number of days from the previous (third dose) BNT162b2 vaccination to the BNT162b2 vaccination received during the study was 129 (87–204) and 165 (117–259) in the coadministration and separate-administration groups, respectively, differing primarily because those in the separate-administration group received BNT162b2 as their visit 2 vaccination.

Randomization and vaccine administration. AE adverse event, SIIV seasonal inactivated influenza vaccine
Immunogenicity
The primary immunogenicity evaluation included 912 (80.4%) participants in the evaluable BNT162b2 immunogenicity population [coadministration group, 87.9% (499/568); separate-administration group, 73.0% (413/566)] and 1016 (89.6%) in the evaluable SIIV immunogenicity population [coadministration group, 91.5% (520/568); separate-administration group, 87.6% (496/566)] (Table S2). The most common reasons for exclusion from the immunogenicity populations were COVID-19 or new SARS-CoV-2 infection after visit 1 and through 1 month after BNT162b2 vaccination in the evaluable BNT162b2 immunogenicity population [15.4% (175/1134)], and not having at least one valid and determinate assay result within 28–42 days after receipt of SIIV in the evaluable SIIV immunogenicity population [10.1% (114/1134)].
Model-adjusted GMRs (95% CI) for coadministration to separate-administration were 0.83 (0.77, 0.89) for full-length S-binding IgG levels and 0.89 (0.77, 1.04), 0.95 (0.83, 1.09), 0.96 (0.85, 1.09), and 1.00 (0.89, 1.13) for the four strain-specific HAI titers (Fig. 3). The prespecified noninferiority criterion was met for full-length S-binding IgG levels and HAI titers for each of the four influenza assay strains (lower bound 95% CI > 0.67). The primary immunogenicity objectives of the study were therefore achieved.

Comparison of coadministration group to separate-administration group of model-adjusted GMRs for the SARS-CoV-2 full-length S-binding IgG levels and influenza strain-specific HAI titers. Data are for the evaluable BNT162b2 immunogenicity population and evaluable SIIV immunogenicity population (defined in Table S1); 95% CIs were based on analysis of logarithmically transformed assay results using a linear regression model that included vaccine group, age group, and corresponding baseline assay results as covariates. Assay results below the LLOQ were set to 0.5 × LLOQ (S-binding IgG and HAI) and results above the ULOQ were set to ULOQ + 1 (HAI only). The dotted line represents the prespecified noninferiority margin. The number of participants with valid and determinate results for the specified assay at both baseline and the given sampling timepoint was 499 in the coadministration group and 413 in the separate-administration group for full-length S-binding IgG levels, and 508–515 and 478–484, respectively, for HAI titers. GMR geometric mean ratio, HAI hemagglutination inhibition assay, IgG immunoglobulin G, LLOQ lower limit of quantitation, S spike protein, SIIV seasonal inactivated influenza vaccine, ULOQ upper limit of quantitation
In an analysis by stratification factors, as expected, participants who were SARS-CoV-2 positive at baseline (positive N-binding antibody result at visit 1 or medical history of SARS-CoV-2 or COVID-19) had higher S-binding GMCs before and after vaccination compared with participants who were SARS-CoV-2 negative at baseline (Fig. S1).
The GMFRs (95% CI) of full-length S-binding IgG levels from baseline (before BNT162b2 vaccination) to 1 month after BNT162b2 vaccination were slightly lower when BNT162b2 was coadministered with SIIV [2.5 (2.4, 2.7)] compared with BNT162b2 administered alone [3.1 (2.9, 3.3); Fig. 4A]. GMFRs of HAI titers from baseline to 1 month after receiving SIIV concomitantly with BNT162b2 ranged from 3.3 to 6.0 across the four influenza strains, and GMFRs after receiving SIIV alone ranged from 3.4 to 6.4 (Fig. 4B).

A GMCs and GMFRs for SARS-CoV-2 full-length S-binding IgG and B GMTs and GMFRs for strain-specific HAI titers from before vaccination to 1 month after vaccination. Data are for the evaluable BNT162b2 immunogenicity population and evaluable SIIV immunogenicity population. Two-sided 95% CIs for GMCs, GMTs, and GMFRs were based on the Student’s t-distribution. In each vaccine group, the number of participants with valid and determinate assay results for the specified assay was 413–499 in the evaluable BNT162b2 immunogenicity population and 478–520 in the evaluable SIIV immunogenicity population. GMC geometric mean concentration, GMFR geometric mean-fold rise, GMT geometric mean titer, HAI hemagglutination inhibition assay, IgG immunoglobulin G, SIIV seasonal inactivated influenza vaccine
Rates of strain-specific HAI titer seroprotection and seroconversion were generally similar across the coadministration and separate-administration groups for each of the four influenza strains (Table S3).
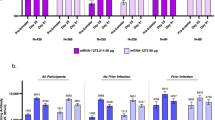
Safety
Local reactions and systemic events within 7 days of vaccination were mostly mild or moderate when BNT162b2 was coadministered with SIIV. Local reactions were assessed only at the BNT162b2 or placebo injection sites. The most commonly reported local reaction, injection-site pain, was much more frequent after receiving BNT162b2 compared with placebo in both the coadministration and separate-administration groups (84.4−86.2% versus 6.6−13.9%; Fig. 5A). Other local reactions were slightly more common after receiving BNT162b2 compared with placebo (4.7−7.6% versus < 1% for redness; 8.9−9.2% versus 0.4−1.1% for swelling). Most local reactions were mild or moderate in severity, with severe local reactions reported in < 0.5% of participants in all vaccine groups, and there were no grade 4 local reactions. Systemic events reported after receiving BNT162b2 and SIIV concomitantly (77.7%) were higher than for those receiving BNT162b2 alone (63.7%), followed by SIIV and placebo (57.2%), or placebo alone (33.8%; Fig. 5B). The most commonly reported systemic events were fatigue (64.0% of participants receiving BNT162b2 and SIIV concomitantly, 50.8% receiving BNT162b2 alone, 42.1% receiving SIIV and placebo, and 21.7% receiving placebo alone) and headache (47.2%, 37.8%, 34.3%, and 20.8%, respectively). Fever (≥ 38.0 °C) was reported infrequently (2.0%, 1.6%, 1.1%, and 1.1%, respectively), and there were no reports of fever > 40.0 °C. The large majority of systemic events were mild or moderate in severity, with severe systemic events reported in < 3.5% of participants across all vaccine groups; there were no grade 4 systemic events. The median onset and duration of local reactions was 1–2 days and 1–3 days, respectively, and 2–5.5 days and 1–2 days for systemic events and was similar across vaccine groups.

A Local reactions and B systemic events reported within 7 days of vaccination. Data are for the safety population. Severity grading of the specific local reactions and systemic events is provided in Table S4. Error bars represent 95% CIs, and numbers above the bars indicate the percentage of participants in each group reporting the specified event. Local reactions were assessed by participants at the BNT162b2 or placebo injection site only. Coadmin (vax 1) = BNT162b2 + SIIV (n = 564); coadmin (vax 2) = placebo (n = 557); separate (vax 1) = SIIV + placebo (n = 562–563); separate (vax 2) = BNT162b2 (n = 553). SIIV seasonal inactivated influenza vaccine
During the 1 month after vaccination, 31.6% of participants reported AEs after receiving BNT162b2 and SIIV concomitantly, 30.5% after receiving SIIV and placebo, 25.1% after receiving BNT162b2 alone, and 29.0% after receiving placebo alone (Fig. 6). AEs were generally similar across the study groups (Table S5). AEs were most commonly reported in the infections and infestations system organ class (in 18.5–21.9% of participants), and were most commonly COVID-19 and upper respiratory tract infection (8.2–11.2% and 3.9–5.5%, respectively, across study groups). AEs considered by the investigator to be related to study vaccine were infrequent and reported by 2.8% of participants after coadministration of BNT162b2 and SIIV, compared with 1.4% after BNT162b2 alone, 1.2% after SIIV and placebo, and 0.9% after placebo alone. The most commonly reported study vaccine-related AEs were lymphadenopathy (BNT162b2 coadministered with SIIV, 0.5%; BNT162b2 alone, 0.7%; SIIV with placebo, 0; placebo alone, 0) and palpitations (BNT162b2 coadministered with SIIV, 0.5%; all other study groups, 0). Two participants experienced AEs leading to study discontinuation (paresthesia in the left lower leg in one participant in the coadministration group 5 days after BNT162b2 and SIIV vaccination, which resolved and was considered related to study vaccine by the investigator; chills, fatigue, pyrexia, myalgia, and tension headaches in one participant starting 1–3 days after receiving placebo with SIIV, which all resolved and were considered related to study vaccine by the investigator). One month after vaccination, SAEs were reported in < 1% of participants after any vaccination; no SAEs were considered by the investigator to be vaccine-related. No cases of myocarditis, pericarditis, or stroke were reported; no severe AEs were considered related to study vaccine; and there were no life-threatening AEs and no deaths.

Adverse events by category reported within 1 month after each vaccination. Data are for the safety population. The numbers above the bars show the percentage of participants who experienced ≥ 1 of the specified type of event after the respective vaccination. AE adverse event, SIIV seasonal inactivated influenza vaccine
Discussion
Vaccination is a critical approach to protect against the severe outcomes of both COVID-19 and influenza, both of which are associated with substantial morbidity and mortality particularly in vulnerable populations [15, 16, 28,29,30]. However, vaccination rates for both COVID-19 and influenza vaccines in many regions are suboptimal [29]. Coadministration of these vaccines, which is currently recommended in several countries and by the WHO [10, 24,25,26], may potentially improve vaccine uptake, and accordingly, vaccination rates, and may therefore maximize protection during peak disease periods and improve the resilience of healthcare systems [28, 29, 31, 32]. Support for this approach as a means to improve vaccination rates comes from an assessment of the 2021 Australian concurrent influenza and COVID-19 vaccination programs [33]. At the time, sequential administration of COVID-19 vaccines with other vaccinations was recommended. However, a substantial decrease in influenza vaccination rates was noted (i.e., 68% by June 2021 compared with 80% in 2020 and 74% in 2019), suggesting that sequential administration may have contributed to decreased influenza vaccination rates.
In this phase 3 study, we observed robust immune responses regardless of concomitant or separate administration of BNT162b2 and SIIV. The immune responses with coadministration of the BNT162b2 COVID-19 vaccine and SIIV were noninferior to the immune responses elicited by either vaccine alone, meeting the prespecified noninferiority criterion for both vaccines. Coadministration of BNT162b2 with SIIV was safe and well tolerated, with an overall safety profile similar to BNT162b2 administered alone. Together, these results provide support for coadministration of BNT162b2 and SIIV.
Determining whether coadministration of COVID-19 and influenza vaccines leads to immune interference is an important consideration when implementing coadministration vaccination strategies [19, 31, 34]. Results from prospective studies assessing coadministration of COVID-19 vaccine booster doses with an influenza vaccine describing the effect on immune responses are available [35,36,37,38,39]. To our knowledge, only one other study has provided a statistical assessment of the noninferiority of the immune response of coadministration of a COVID-19 vaccine and an influenza vaccine compared with separate administration [38]. In that randomized study, immunogenicity of coadministration of a BNT162b2 booster with an influenza vaccine was compared with BNT162b2 alone in 154 participants 60 years and older who were fully vaccinated with any COVID-19 vaccine [38]. That study failed to confirm the noninferiority of concurrent administration compared with administration of BNT162b2 alone, and both the levels and functionality of the immune response against SARS-CoV-2 were decreased with coadministration, suggesting possible immune interference as a result of coadministration. A key limitation of that study was the heterogeneous history of prior COVID-19 vaccinations across vaccine platforms (viral vectored, mRNA) that likely contributed to more variable baseline immunity that could impact the ability to achieve noninferiority with subsequent coadministration of COVID-19 vaccine with a seasonal influenza vaccine.
The tolerability or safety profile of either vaccine is another important consideration in assessing coadministration approaches [31]. Prospective studies have shown that tolerability and safety profiles do not appear to be adversely affected with coadministration of a COVID-19 vaccine with influenza vaccine [35,36,37,38,39]. In a phase 4 study that included 679 participants who were randomized to receive a second dose of BNT162b2 or the ChAdOx1 adenovirus COVID-19 vaccine, at the same time as one of three different influenza vaccines, no safety concerns with coadministration were identified [35]. Support for the safety of coadministration of mRNA COVID-19 vaccines with SIIV is also available from an analysis of data from the US Vaccine Adverse Event Reporting System (VAERS) safety surveillance program [40]. The analysis of data from July 2021 to June 2022 concluded that no new or unexpected safety issues were identified, and the safety profile of coadministration was generally consistent with the safety profile of either vaccine given alone. Additionally, a surveillance study from the US Centers for Disease Control and Prevention (CDC) found that systemic events occurred 8–11% more frequently in individuals who received mRNA COVID-19 vaccine booster doses with seasonal influenza vaccination (n = 92,023) than in those who received the COVID-19 vaccine booster alone (n = 889,076) [41]. However, events reported in that surveillance study were usually mild. Finally, a preliminary potential ischemic stroke signal was identified by the CDC for the Omicron BA.4/BA.5 variant-adapted bivalent BNT162b2 vaccine among older adults [42]. However, further analyses of the Omicron BA.1 variant-adapted bivalent BNT162b2 vaccine given alone or given on the same day as influenza vaccination identified no increased risk of ischemic or hemorrhagic stroke, including among older adults [43]. With the accumulation of further safety data, the initial CDC finding has decreased and it is thought that factors other than vaccination contributed to this preliminary finding [42], and the CDC continues to recommend coadministration of COVID-19 vaccines with influenza vaccines [44]. Additionally, we saw no stroke events in our study, which used the original BNT162b2 vaccine.
Strengths of our study include the randomized design and the assessment of noninferiority of the immune response of coadministration of BNT162b2 and SIIV compared with separate administration of either vaccine, while allowing all participants to receive both vaccines. Limitations of our study should also be noted. The study was conducted in Australia and New Zealand in 18- to 64-year-old adults who had received three prior doses of BNT162b2; however, we anticipate that the findings will be generalizable to other adult populations, such as the elderly, and across geographic regions and among individuals who may have received other COVID-19 vaccines for their primary series. The follow-up for safety was also limited to 1 month after each vaccination. The primary BNT162b2 immunogenicity endpoint of our study was assessment of full-length S-binding IgG levels. It is anticipated that subsequent analysis of SARS-CoV-2 neutralizing titers will be supportive of the findings of the primary analysis because trends for S-binding IgG and neutralizing responses have been generally similar following COVID-19 vaccination. Notably, the only other study powered to assess noninferiority of coadministration of a COVID-19 vaccine and influenza vaccine compared with COVID-19 booster vaccination alone also assessed IgG responses [38]. Additionally, Omicron-adapted vaccines are now being used in several countries, including Australia, New Zealand, and the USA [10, 11, 45]. However, we anticipate that the findings reported here for original BNT162b2 will be generalizable to the variant-adapted vaccines, and regulators are not generally requiring further clinical data with every update to the COVID-19 mRNA vaccine platform, nor with every seasonal update to SIIVs [46, 47]. Finally, the effectiveness of coadministration vaccination strategies to protect against COVID-19 and influenza illness remains to be determined, and such confirmation would require real-world studies.
In conclusion, this study demonstrated that BNT162b2 can be administered concomitantly with SIIV in adults 18–64 years of age. BNT162b2 was safe and well tolerated when coadministered with SIIV and showed robust immune responses that were noninferior to separate administration of BNT162b2 and SIIV. Acceptance of influenza vaccine and COVID-19 vaccine coadministration is an important consideration, with studies reporting low public acceptance of this approach, particularly attributed to concerns regarding decreased effectiveness and increased adverse reactions with coadministration [31]. The rigorous clinical trial data reported here supporting the coadministration of these vaccines should help to assuage these concerns.
Data Availability
Upon request, and subject to review, Pfizer will provide the data that support the findings of this study. Subject to certain criteria, conditions and exceptions, Pfizer may also provide access to the related individual de-identified participant data. See https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information.
References
Polack FP, Thomas SJ, Kitchin N, et al. Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine. N Engl J Med. 2020;383:2603–15.
Moreira ED Jr, Kitchin N, Xu X, et al. Safety and efficacy of a third dose of BNT162b2 Covid-19 vaccine. N Engl J Med. 2022;386:1910–21.
Bar-On YM, Goldberg Y, Mandel M, et al. Protection of BNT162b2 vaccine booster against Covid-19 in Israel. N Engl J Med. 2021;385:1393–400.
Feikin DR, Higdon MM, Abu-Raddad LJ, et al. Duration of effectiveness of vaccines against SARS-CoV-2 infection and COVID-19 disease: results of a systematic review and meta-regression. Lancet. 2022;399:924–44.
Surie D, DeCuir J, Zhu Y, et al. Early estimates of bivalent mRNA vaccine effectiveness in preventing COVID-19-associated hospitalization among immunocompetent adults aged ≥ 65 years—IVY network, 18 States, September 8-November 30, 2022. MMWR Morb Mortal Wkly Rep. 2022;71:1625–30.
Tenforde MW, Weber ZA, Natarajan K, et al. Early estimates of bivalent mRNA vaccine effectiveness in preventing COVID-19-associated emergency department or urgent care encounters and hospitalizations among immunocompetent adults—VISION network, Nine States, September-November 2022. MMWR Morb Mortal Wkly Rep. 2022;71:1616–24.
Link-Gelles R, Weber ZA, Reese SE, et al. Estimates of bivalent mRNA vaccine durability in preventing COVID-19-associated hospitalization and critical illness among adults with and without immunocompromising conditions—VISION Network, September 2022-April 2023. MMWR Morb Mortal Wkly Rep. 2023;72:579–88.
Zheng Y, Wang Y. How seasonality and control measures jointly determine the multistage waves of the COVID-19 epidemic: a modelling study and implications. Int J Environ Res Public Health. 2022;19:6404.
Wiemken TL, Khan F, Puzniak L, et al. Seasonal trends in COVID-19 cases, hospitalizations, and mortality in the United States and Europe. Sci Rep. 2023;13:3886.
Australian Government Department of Health and Aged Care. Clinical recommendations for COVID-19 vaccines. https://www.health.gov.au/our-work/covid-19-vaccines/advice-for-providers/clinical-guidance/clinical-recommendations. Accessed 23 May 2023.
New Zealand Ministry of Health. COVID-19 vaccine: Boosters. https://www.health.govt.nz/our-work/diseases-and-conditions/covid-19-novel-coronavirus/covid-19-vaccines/covid-19-vaccine-boosters. Accessed 23 May 2023.
Rosenblum HG, Wallace M, Godfrey M, et al. Interim recommendations from the Advisory Committee on Immunization Practices for the use of bivalent booster doses of COVID-19 vaccines—United States, October 2022. MMWR Morb Mortal Wkly Rep. 2022;71:1436–41.
Danish Health Authority. Vaccination. https://www.sst.dk/en/english/Corona-eng/Guidelines-vaccination-and-disease-prevention/Vaccination. Accessed 15 June 2023.
European Centre for Disease Prevention and Control. Interim public health considerations for COVID-19 vaccination roll-out during 2023. https://www.ecdc.europa.eu/en/publications-data/interim-public-health-considerations-covid-19-vaccination-roll-out-during-2023. Accessed 15 June 2023.
Macias AE, McElhaney JE, Chaves SS, et al. The disease burden of influenza beyond respiratory illness. Vaccine. 2021;39:A6–A14.
Grohskopf LA, Blanton LH, Ferdinands JM, et al. Prevention and control of seasonal influenza with vaccines: recommendations of the Advisory Committee on Immunization Practices—United States, 2022–23 influenza season. MMWR Recomm Rep. 2022;71:1–28.
Swets MC, Russell CD, Harrison EM, et al. SARS-CoV-2 co-infection with influenza viruses, respiratory syncytial virus, or adenoviruses. Lancet. 2022;399:1463–4.
Pawlowski C, Silvert E, O’Horo JC, et al. SARS-CoV-2 and influenza coinfection throughout the COVID-19 pandemic: an assessment of coinfection rates, cohort characteristics, and clinical outcomes. PNAS Nexus. 2022;1:pgac071.
Altmann DM, Boyton RJ. Vaccine efficacy and immune interference: co-administering COVID-19 and influenza vaccines. Lancet Respir Med. 2022;10:125–6.
Krumbein H, Kümmel LS, Fragkou PC, et al. Respiratory viral co-infections in patients with COVID-19 and associated outcomes: a systematic review and meta-analysis. Rev Med Virol. 2023;33:e2365.
Cong B, Deng S, Wang X, Li Y. The role of respiratory co-infection with influenza or respiratory syncytial virus in the clinical severity of COVID-19 patients: a systematic review and meta-analysis. J Glob Health. 2022;12:05040.
Kim H, Webster RG, Webby RJ. Influenza virus: dealing with a drifting and shifting pathogen. Viral Immunol. 2018;31:174–83.
Australian Government Department of Health and Aged Care. Influenza (flu) vaccine (17 April 2023). https://www.health.gov.au/topics/immunisation/vaccines/influenza-flu-vaccine. Accessed 11 May 2023.
The Immunisation Advisory Centre (Te Whatu Ora Health New Zealand). FLU 2023 essential information for health professionals. https://assets.website-files.com/61f35aaad08e65b4f5a55423/644eeef5afc045a87db16291_IMAC_Flu_Kit_27-04-2023.pdf. Accessed 11 May 2023.
US Centers for Disease Control and Prevention. Interim clinical considerations for use of COVID-19 vaccines currently approved or authorized in the United States. https://www.cdc.gov/vaccines/covid-19/clinical-considerations/interim-considerations-us.html. Accessed 11 May 2023.
World Health Organization. Coadministration of seasonal inactivated influenza and COVID-19 vaccines. https://www.who.int/publications/i/item/WHO-2019-nCoV-vaccines-SAGE_recommendation-coadministration-influenza-vaccines. Accessed 23 May 2023.
European Commission. Joint Statement by Commissioner Stella Kyriakides, WHO Regional Director for Europe, Dr Hans Henri P. Kluge and Director of the ECDC Dr Andrea Ammon: working together towards COVID-19 and seasonal influenza vaccinations for this winter; 2022.
Bonanni P, Steffen R, Schelling J, et al. Vaccine co-administration in adults: an effective way to improve vaccination coverage. Hum Vaccin Immunother. 2023;19:2195786.
Domnich A, Orsi A, Trombetta CS, et al. COVID-19 and seasonal influenza vaccination: cross-protection, co-administration, combination vaccines, and hesitancy. Pharmaceuticals (Basel). 2022;15:322.
Zhang JJ, Dong X, Liu GH, Gao YD. Risk and protective factors for COVID-19 morbidity, severity, and mortality. Clin Rev Allergy Immunol. 2023;64:90–107.
Domnich A, Grassi R, Fallani E, et al. Acceptance of COVID-19 and influenza vaccine co-administration: insights from a representative Italian survey. J Pers Med. 2022;12:139.
Venuto R, Giunta I, Cortese R, et al. The importance of COVID-19/influenza vaccines co-administration: an essential public health tool. Infect Dis Rep. 2022;14:987–95.
Van Buynder PG, Newbound A, MacIntyre CR, et al. Australian experience of the SH21 flu vaccination program during the COVID-19 vaccine program. Hum Vaccin Immunother. 2021;17:4611–6.
Xie Y, Tian X, Zhang X, Yao H, Wu N. Immune interference in effectiveness of influenza and COVID-19 vaccination. Front Immunol. 2023;14:1167214.
Lazarus R, Baos S, Cappel-Porter H, et al. Safety and immunogenicity of concomitant administration of COVID-19 vaccines (ChAdOx1 or BNT162b2) with seasonal influenza vaccines in adults in the UK (ComFluCOV): a multicentre, randomised, controlled, phase 4 trial. Lancet. 2021;398:2277–87.
Toback S, Galiza E, Cosgrove C, et al. Safety, immunogenicity, and efficacy of a COVID-19 vaccine (NVX-CoV2373) co-administered with seasonal influenza vaccines: an exploratory substudy of a randomised, observer-blinded, placebo-controlled, phase 3 trial. Lancet Respir Med. 2022;10:167–79.
Izikson R, Brune D, Bolduc JS, et al. Safety and immunogenicity of a high-dose quadrivalent influenza vaccine administered concomitantly with a third dose of the mRNA-1273 SARS-CoV-2 vaccine in adults aged ≥65 years: a phase 2, randomised, open-label study. Lancet Respir Med. 2022;10:392–402.
Dulfer EA, Geckin B, Taks EJM, et al. Timing and sequence of vaccination against COVID-19 and influenza (TACTIC): a single-blind, placebo-controlled randomized clinical trial. Lancet Reg Health Eur. 2023;29:100628.
Radner H, Sieghart D, Jorda A, et al. Reduced immunogenicity of BNT162b2 booster vaccination in combination with a tetravalent influenza vaccination: results of a prospective cohort study in 838 health workers. Clin Microbiol Infect. 2023;29:635–41.
Moro PL, Zhang B, Ennulat C, et al. Safety of co-administration of mRNA COVID-19 and seasonal inactivated influenza vaccines in the Vaccine Adverse Event Reporting System (VAERS) during July 1, 2021-June 30, 2022. Vaccine. 2023;41:1859–63.
Hause AM, Zhang B, Yue X, et al. Reactogenicity of simultaneous COVID-19 mRNA booster and influenza vaccination in the US. JAMA Netw Open. 2022;5:e2222241.
US Food and Drug Administration. CDC and FDA Identify Preliminary COVID-19 Vaccine Safety Signal for Persons Aged 65 Years and Older. https://www.fda.gov/vaccines-blood-biologics/safety-availability-biologics/cdc-and-fda-identify-preliminary-covid-19-vaccine-safety-signal-persons-aged-65-years-and-older. Accessed 29 June 2023.
Andrews N, Stowe J, Miller E, Ramsay M. BA.1 bivalent COVID-19 vaccine use and stroke in England. JAMA. 2023;330:184–5.
US Centers for Disease Control and Prevention. Interim Clinical Considerations for Use of COVID-19 Vaccines Currently Authorized in the United States. https://www.cdc.gov/vaccines/covid-19/clinical-considerations/interim-considerations-us.html. Accessed 11 May 2023.
US Food and Drug Administration. Coronavirus (COVID-19) update: FDA authorizes changes to simplify use of bivalent mRNA COVID-19 vaccines; 2023. https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-changes-changessimplify-use-bivalent-mrna-covid-19-vaccines#:~:text=Today%2C%20the%20U.S.%20Food%20and,4%2FBA. Accessed 8 Sept 2023.
European Medicines Agency. ECDC-EMA statement on updating COVID-19 vaccines composition for new SARS-CoV-2 virus variants; 2023. https://www.ecdc.europa.eu/en/news-events/ecdc-ema-statement-updating-covid-19-vaccines-composition-newsars-cov-2-virus-variants. Accessed 8 Sept 2023.
World Health Organization. Statement on the antigen composition of COVID-19 vaccines. https://www.who.int/news/item/18-05-2023-statement-on-the-antigen-composition-of-covid-19-vaccines. Accessed 16 June 2023.
Acknowledgements
We thank all the participants who volunteered for this trial; the investigators and study site personnel in the C4591030 Clinical Trial Group for their contributions; the independent Data Monitoring Committee (Jonathan Zenilman, Kathryn Edwards, Robert Belshe, Lawrence Stanberry, Steve Self, Robert Philips Heine, and Heather S. Lipkind) for their review of the trial safety data; the following Pfizer staff: Vrunda Parikh and Vishva Raj Bangad, as well as Pfizer colleagues from Vaccine Clinical Research and Development, High-Throughput Clinical Testing, Clinical Development Operations, Global Site and Study Operations, Pharmaceutical Sciences, Vaccine Regulatory, and Project Management for their contributions to the success of this study; Richard De Solom (Pfizer colleague at the time this study was completed); and the following BioNTech colleagues: Nadine Salisch, Shon Remich, and Ruben Rizzi.
We thank the investigators for this trial: Mark Arya, Eugene Athan, Timothy Blackmore, Sheetal Bull, Andrew Edwards, Emma Esquilant, Joanne Finlay, Paul Hamilton, Tiwini Hemi, Timothy Humphrey, Jackie Kamerbeek, Jane Kerr, Jen Kok, Anthony McGirr, Barnaby Montgomery, Louise Murdoch, A. Munro Neville, Dean Quinn, Davitt Sheahan, Susan Smith, Richard Stubbs, Maelen Tagelagi, Claire Thurlow, Michael Williams, and Joanna Wojciechowska.
Medical Writing/Editorial Assistance
Tricia Newell, PhD, and Sheena Hunt, PhD, of ICON (Blue Bell, PA, USA) provided medical writing and editorial support funded by Pfizer.
Funding
This study was sponsored by BioNTech and funded by Pfizer Inc and BioNTech. Pfizer Inc is funding the journal’s Rapid Service fee.
Author information
Authors and Affiliations
Consortia
Contributions
Louise Murdoch and Claire Lu were involved in data collection. Karen Quan, James A. Baber, Agnes W.Y. Ho, Ying Zhang, Xia Xu, Claire Lu, David Cooper, Kenneth Koury, Stephen P. Lockhart, Annaliesa S. Anderson, Özlem Türeci, Uğur Şahin, Kena A. Swanson, William C. Gruber, and Nicholas Kitchin contributed to either study conception and design and/or data analysis. Louise Murdoch, Karen Quan, James A. Baber, Agnes W.Y. Ho, Ying Zhang, Xia Xu, Claire Lu, David Cooper, Kenneth Koury, Stephen P. Lockhart, Annaliesa S. Anderson, Özlem Türeci, Uğur Şahin, Kena A. Swanson, William C. Gruber, and Nicholas Kitchin commented on all draft versions of the manuscript and read and approved the final manuscript for submission.
Corresponding author
Ethics declarations
Conflict of Interest
Louise Murdoch has no conflict of interest involving Pfizer or BioNTech to declare. Karen Quan, James A. Baber, Agnes W.Y. Ho, Ying Zhang, Xia Xu, Claire Lu, David Cooper, Kenneth Koury, Stephen P. Lockhart, Annaliesa S. Anderson, Kena A. Swanson, William C. Gruber, and Nicholas Kitchin are employees of Pfizer and may hold stock or stock options. Özlem Türeci and Uğur Şahin are employees of BioNTech and may hold stock or stock options.
Ethical Approval
The study was conducted in accordance with the consensus ethical principles derived from international guidelines, including the Declaration of Helsinki and Council for International Organizations of Medical Sciences International Ethical Guidelines, as well as applicable International Council for Harmonisation Good Clinical Practice guidelines, laws, and regulations, including privacy laws. The ethics committees for this study were Monash Health Human Research Ethics Committee (Australia) and the COVID-19 Emergency Standard Operating Procedures of the Northern B Health and Disability Ethics Committee (New Zealand). All participants who received study vaccinations provided written informed consent.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Murdoch, L., Quan, K., Baber, J.A. et al. Safety and Immunogenicity of the BNT162b2 Vaccine Coadministered with Seasonal Inactivated Influenza Vaccine in Adults. Infect Dis Ther 12, 2241–2258 (2023). https://doi.org/10.1007/s40121-023-00863-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40121-023-00863-5