
Abstract
Introduction
Dosing frequency is an important factor influencing medication compliance in patients with heart failure (HF), which in turn is imperative in achieving the desired therapeutic outcome. Here we assessed the efficacy and safety of ivabradine prolonged-release (PR) once-daily (test) vs. ivabradine immediate-release (IR) twice-daily (reference) formulations in patients with stable chronic HF with systolic dysfunction.
Methods
Patients with sinus rhythm and heart rate (HR) ≥ 50 bpm, left ventricular ejection fraction ≤ 40% (HF with reduced ejection fraction), on guideline-based standard care, receiving a stable dose of ivabradine IR 5/7.5 mg twice daily for ≥ 1 month were enrolled in this randomized, double-blind, phase 3 non-inferiority study. Patients were randomly assigned 1:1 to ivabradine PR (10 mg/15 mg) based on the ivabradine IR dosage or continued ivabradine IR (5 mg/7.5 mg). The primary endpoint was change in resting ECG HR from baseline to the end of 3 months, assessed by 12-lead ECG. Safety assessments and 24-h Holter HR monitoring (in a subgroup of patients) were also performed. Non-inferiority was concluded if the upper limit of the 95% CI of the difference between the test and reference was less than the margin of 6.5 bpm in the per-protocol set.
Results
A total of 169 out of 180 randomized patients (93.9%) completed the study (PR = 84; IR = 85). The least-square mean (standard error [SE]) for change in HR from baseline to 3 months was 0.76 (1.188; 95% CI −1.59:3.11) in ivabradine PR vs. ivabradine IR, which was within the pre-specified margin of 6.5 bpm, confirming the non-inferiority of ivabradine PR. The change from baseline to 3 months was comparable between the treatment groups for 24-h Holter ECG monitoring (p = 0.3701), mean HR awake (p = 0.3423), and mean HR asleep (p = 0.1501). Thirty-nine treatment-emergent adverse events (TEAEs) were reported; the majority in both groups were of mild or moderate severity and were subsequently resolved. Seven serious adverse events were reported (ivabradine PR = 2; ivabradine IR = 5), of which one was fatal (ivabradine IR group). The bradycardia events reported were comparable between groups.
Conclusion
Ivabradine PR was found to be non-inferior to ivabradine IR in the management of patients with stable CHF, with a comparable safety profile. Once-daily ivabradine PR effectively maintained the HR in patients shifted from the ivabradine IR twice-daily regimen, and thus may aid in improving treatment compliance.
Trial Registration
CTRI/2018/04/013464 (Trial Registered Prospectively on 24/04/2018)
Similar content being viewed by others


Why carry out this study? |
Heart failure management involves multidrug treatment with different dosage regimens. |
Dosing frequency is an important factor influencing medication compliance in patients with heart failure. |
The PROFICIENT study was designed to assess the non-inferiority of the new ivabradine prolonged-release (PR) once-daily formulation compared with the conventional immediate-release (IR) twice-daily formulation with regard to the efficacy and safety parameters in patients with stable chronic heart failure with systolic dysfunction. |
What was learned from the study? |
The once-daily PR formulation of ivabradine was found to be non-inferior to twice-daily ivabradine IR, when added to background guideline-based medical treatment, in patients with stable chronic heart failure. |
The safety profile was comparable between the treatment groups. |
Change in heart rate was comparable between the treatment groups as measured by 24-hr Holter monitoring (mean 24-hr HR, mean awake HR and mean asleep HR). |
Once-daily ivabradine PR effectively maintained the heart rate in patients shifted from the ivabradine IR twice-daily regimen, and thus may aid in improving treatment compliance. |
Digital Features
This article is published with digital features to facilitate understanding of the article. You can access the digital features on the article’s associated Figshare page. To view digital features for this article go to https://doi.org/10.6084/m9.figshare.12932993.
Introduction
Heart failure (HF) is rapidly emerging as a global health issue, with an estimated prevalence of > 37.7 million individuals [1]. It is often associated with shorter life expectancy and poor quality of life, and imposes a substantial burden on the health-care system. The prevalence of HF in India is also on the rise, as it remains doubly burdened by increased risk factors for traditional cardiovascular disease (CVD) and persistence of pre-transitional diseases such as rheumatic heart disease, untreated congenital heart disease, endomyocardial fibrosis, and anemia [2]. The current estimates of the incidence of HF in India vary widely from 1.3 to 23 million [3].
Angiotensin-converting enzyme inhibitors/angiotensin receptor antagonists, β-blockers, angiotensin II receptor blocker-neprilysin inhibitors, and mineralocorticoid receptor antagonists are the mainstay of therapy for HF with reduced ejection fraction (HFrEF; EF < 40%), which accounts for 60% of HF cases in adults. These agents play a fundamental role in modifying the course of systolic HF and are widely used in conjunction with diuretics to alleviate symptoms and signs of congestion [4,5,6,7,8].
However, despite clear guideline recommendations, target doses of these important therapies are often underused in clinical practice. This may be attributed to the increased risk of adverse reactions such as low blood pressure and renal dysfunction, in addition to negative chronotropism associated with the up-titration of these agents in response to persistently elevated heart rate (HR) [9]. Moreover, β-blockers may have other undesired actions on the heart, including an effect on myocardial contractility, in addition to their attenuating impact on HR [10].
Ivabradine continues to be an important therapy to achieve desirable HR in patients with HF since its US Food and Drug Administration (FDA) licensing in 2015 and European Medicines Agency (EMA) approval in 2012 [11]. Ivabradine exerts an inhibitory effect on the If of the sinoatrial node by reducing its diastolic depolarization slope, leading to a reduction in HR without affecting myocardial contractility or relaxation, ventricular repolarization, or intracardiac conduction. At a dose that induces comparable reductions in HR, β-blockers may depress left ventricular (LV) contractile function and increase diastolic time, whereas ivabradine does not affect maximal LV pressure increase (LV dP/dtmax) but increases diastolic time to a greater extent [12].
Several placebo-controlled, multicenter clinical studies have shown the effect of ivabradine in the treatment of a variety of CVD including coronary artery disease (CAD) with LV systolic dysfunction, chronic HF (CHF), and CAD without clinical HF [10, 13,14,15,16]. The recent J-SHIFT study showed that systolic blood pressure remained unchanged when the HR was reduced with ivabradine. This maintenance of hemodynamics during HR reduction is essential to achieving future cardiac reverse remodeling and better survival [17, 18].
Compliance with the medical regimen is important in the optimal management of HF, and medication dosing frequency has an important effect on medication adherence. According to the World Health Organization, only 50% of chronically ill patients adhere to their prescribed medications in developed countries, and this rate is lower in developing countries [19, 20]. Patients with better medication compliance have reported greater treatment effectiveness and fewer HF symptoms and subsequent consequences such as hospitalizations and death [21]. Moreover, patients were found to be more compliant with once-daily compared with twice-daily or thrice-daily treatment regimens [22, 23].
Ivabradine is currently available as immediate-release (IR) tablets in strengths of 2.5 mg, 5 mg, and 7.5 mg, for twice-daily dosing. A prolonged-release (PR) formulation with once-daily dosing compared with the currently available twice-daily IR formulation may help not only in decreasing fluctuations in plasma drug concentrations (results presented elsewhere) and achieving the desired therapeutic objective, but also in increasing patient compliance.
Hence we designed the PROlonged Release Formulation of Ivabradine OnCe-DaIly in HEart Rate ManagemeNT (PROFICIENT) study to assess the non-inferiority of the new once-daily ivabradine PR compared with the conventional twice-daily IR formulation in the efficacy (change in HR from baseline to 3 months, assessed by 12-lead electrocardiogram [ECG] recordings) and safety parameters in patients with stable chronic HF with systolic dysfunction. The 24-h ambulatory HR with Holter monitoring was also assessed in a subgroup of patients.
Methods
Patients
Patients (men and women aged 18–70 years, inclusive) with systolic HF, New York Heart Association (NYHA) functional class II or III symptoms for ≥ 6 weeks, on guideline-based standard care for stable CHF [4], receiving a stable dose of ivabradine IR 5 mg/7.5 mg twice daily for ≥ 1 month were enrolled in the PROFICIENT study. Patients had to be in ECG-documented sinus rhythm with resting HR (≥ 50 beats per minute [bpm.]) and left ventricular ejection fraction (LVEF) ≤ 40% and willing to participate in the study.
Major exclusion criteria were as follows: recent myocardial infarction, coronary revascularization, stroke, or transient ischemic attack (≤ 3 months), or with a transplanted heart, implanted pacemaker, implantable cardioverter defibrillator, or cardiac resynchronization therapy; permanent atrial fibrillation or flutter or any other cardiac arrhythmias which could interfere with the function of the sinoatrial node; sick sinus syndrome, sinoatrial block, congenital long QT or treatment with QT-prolonging medications, second degree and complete atrioventricular block; stroke or transient cerebral ischemia within previous ≤ 3 months prior to screening; unstable or acute HF, unstable angina; severe or uncontrolled hypertension (systolic blood pressure [SBP] ≥ 190 mmHg or diastolic BP [DBP] ≥ 110 mmHg), uncontrolled hypotension (SBP ≤ 90 mmHg or DBP ≤ 50 mmHg); hospitalization for worsening HF (NYHA class II or higher) ≤ 3 months prior to screening; or scheduled for coronary revascularization, or likely to require surgery for valvular disease during the study period. Pregnant or lactating women, or patients with any condition that did not justify their participation in the study as judged by the investigator; with any contraindication to the study drug or its excipients; or who had participated in any other clinical study in the past 3 months before screening were excluded from the study.
The study protocol and amendments were approved by the respective institutional ethics committee of each site (full list of institutional review boards [IRBs] is included as supplementary material). The study was carried out per the protocol and in compliance with the International Council for Harmonisation Guideline for Good Clinical Practice, Declaration of Helsinki, Schedule Y (amended version, 2013) of the Central Drugs Standard Control Organization, Ministry of Health and Family Welfare, Government of India, and ethical guidelines for biomedical research on human participants, Indian Council of Medical Research. Written informed consent was obtained from all patients before enrollment. Additional consent was obtained from patients who participated in the 24-h Holter ECG monitoring subgroup analysis. This study is registered at Clinical Trials Registry—India (CTRI/2018/04/013464 [trial registered prospectively on 24/04/2018]).
Study Design, Randomization, and Blinding
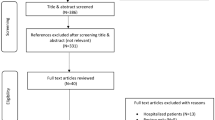
PROFICIENT was a randomized, double-blind, double-dummy, multicenter, phase 3 non-inferiority study conducted from May 2018 through April 2019 and included patients from 21 centers across India (Appendix 1). The study consisted of six visits (Fig. 1).

CONSORT flow diagram of study design
Upon confirming the eligibility of inclusion at the screening visit (visit 1), ivabradine IR 5 mg or 7.5 mg (Coralan®, Servier Laboratories, France) was administered to all patients during the run-in period (8 days; visit 2). The eligible patients were randomly allocated (day 0; visit 3) to treatment groups (1:1 ratio) using a centralized interactive web response system, to receive either once-daily ivabradine PR (Abbott Healthcare Pvt Ltd, India), 10 mg or 15 mg, or twice-daily ivabradine IR 5 mg or 7.5 mg, based on their baseline ivabradine doses of 5 mg or 7.5 mg twice daily.
The patients took the first dose on the day following randomization. A simple randomization technique with a unique number was followed. During the treatment period (day 16 to day 90), patients continued the assigned study treatment. The end-of-study (EOS) visit (visit 6; day 91) followed the last dose on the 90th day.
The test (ivabradine PR) and the reference product (ivabradine IR) differed in appearance, shape, smell, taste, packaging, and dosing regimen; hence, to maintain the blinding, a double-dummy approach was followed. Each patient was administered an identical number of tablets (i.e. four tablets) daily. This ensured that the patients randomized to either of the dosing regimens (test or reference) received at least one placebo tablet at each dosing. Patients randomized to the ivabradine PR group received two placebo tablets during any one dosing. The study drug administrator was trained in drug handling, administration, accountability, storage, and medication allocation. Patients who were willing to participate in the 24-h ECG Holter subgroup analysis underwent 24-h Holter ECG monitoring on day 1 and day 90. A patient diary was provided to all patients for recording dosing details to monitor compliance.
The PROFICIENT trial committee was responsible for the clinical and scientific conduct of the study.
Outcomes
Efficacy
The primary endpoint was the change in resting HR from baseline to the end of 3 months between the ivabradine PR and ivabradine IR groups. The resting HR was measured in beats per minute (bpm) by automated, calibrated, standard 12-lead ECG [RMS Enterprises, Recorders and Medicare Systems (P) Ltd.] supplied to all the sites. The mean resting HR was measured at each visit based on three consecutive ECG recordings, within 30 min after 10 min of initial rest in the supine position. Analysis of the 24-h ECG recordings was performed using the instrument software yielding HR (bpm), PQ/PR interval (ms), QRS interval (ms), QT interval (ms), RR interval (ms), and QTc interval (ms) [all machine-read]. The machine-read QTc interval was calculated using Bazett’s/Fridericia’s formula.
24-Hour Holter ECG Monitoring
The 24-h average HR was also assessed using Holter ECG monitoring (awake and sleep). The continuous recordings were obtained throughout a 24-h period before randomization and visit 6 (day 91) in a subset of 50 patients. All Holter recordings were read using the dedicated RMS system [RMS Enterprises, Recorders, and Medicare Systems (P). Ltd]. The 24-h Holter ECG recordings were analyzed by experienced Holter technicians and cardiologists who were blinded to study treatment allocation and to temporal sequences of recording.
Safety
Safety outcomes included the incidence and severity of adverse events (AE), serious adverse events (SAEs), and treatment-emergent adverse events (TEAEs). The incidence of hospitalizations for worsening HF, other CV reasons, and mortality rates were also assessed at the end of 3 months, compared to baseline.
Covariates
Demographic characteristics, laboratory parameters (hematology, blood biochemistry), vital signs, medical history, concomitant medication, self-reported smoking, and alcohol consumption status were assessed.
The clinical significance of the ECG, safety, comorbid conditions, and hematology/biochemistry parameters were assessed by the investigator or designated personnel, and the abnormality based on examination or laboratory values, if any, was specified.
Statistical Methods
The safety analysis set consisted of all patients who received at least one dose of study medications. The full analysis set included all patients in the safety analysis who had data for at least one post-baseline assessment of any efficacy measurement. The per-protocol analysis set consisted of all patients who were included in the FA analysis set and did not present any major protocol violation. The Holter group set consisted of all patients who consented and for whom 24-h Holter ECG monitoring was performed.
Considering the active drug in both treatment groups, the assumption was that the HR from baseline to end of treatment would be maintained within the range of values defined for 5 mg or 7.5 mg ivabradine IR (> 50 and ≤ 75). For the same, a sample size of 72 was estimated assuming an absolute inferiority margin of 6.5 bpm, standard deviation (SD) of 12, and effect size of 0.54, with 90% power to establish non-inferiority between the groups using the one-sided test at a 95% confidence interval (CI). An additional 18 patients per group were enrolled considering a dropout rate of 20%.
All analyses were carried out as two-sided on a 5% level of significance and 95% CI. Analysis of covariance (ANCOVA) with treatment as factor and baseline as a covariate was used for treatment comparison. Continuous variables are summarized using mean (SD) and categorical variables as number (percentage) of patients within each level. The primary objective was to test the non-inferiority of ivabradine PR compared with the ivabradine IR tablet by the non-inferiority absolute margin (δ) of 6.5 bpm, at one-sided alpha (α) of 0.025. Non-inferiority was concluded if the upper limit of the 95% CI of the difference for mean change in HR from baseline to 3 months between the PR and IR groups was less than the pre-specified margin of 6.5 bpm. The Holter subgroup analysis was performed similarly to the primary analysis but was not statistically powered. The change in HR (measured by automated 12-lead ECG) vs. HR measured by 24-h Holter ECG (24-h, awake, and sleep) was assessed as an exploratory endpoint to understand the trends if any. The statistical analysis was carried out using the most recent SAS® (Statistical Analysis Software) system version 9.4. (SAS Institute Inc., Cary, NC, USA).
Results
Of the 188 patients enrolled, 180 (95.7%) were randomized (1:1) to either the ivabradine PR (n = 90) or ivabradine IR (n = 90) group. The safety population included 179 (99.4%) patients (89 in the ivabradine PR group and 90 in the IR group), and the per-protocol analysis set comprised 160 patients (80 in the ivabradine PR group and 80 in the IR group). A total of 169 patients (93.9%) completed the study (Fig. 1). The most common reason for study non-eligibility or non-randomization was run-in failure.
Demographic and baseline characteristics were well-balanced between the groups (Table 1). The mean ± SD age of the study population was 54.0 ± 10.02 years. The HF was of ischemic cause in 143 (79.9%) patients. A total of 24.6% of the study population had previous congestive cardiac failure, and 15.1% had previous myocardial infarction.
All patients were on a stable dose of ivabradine IR 5 mg or 7.5 mg twice daily for more than 1 month. A total of 169 patients were receiving ivabradine 5 mg, and ten patients were receiving 7.5 mg. Post-randomization, 83 (93.3%) patients were on ivabradine PR 10 mg once daily, and 86 (95.6%) patients were on ivabradine IR 5 mg twice daily, while 6 (6.7%) and 4 (4.4%) patients were on 15 mg PR once daily and 7.5 mg IR twice daily, respectively. The most commonly used concomitant medications were diuretics (147 [82.1%]), β-blockers (131 [73.2%]), lipid-modifying agents (131 [73.2%]), and agents acting on the renin–angiotensin system (122 [68.2%]) (Table 2).
Efficacy
The mean HR at baseline was 62.8 ± 9.47 bpm in the ivabradine PR group and 63.6 ± 8.85 bpm in the ivabradine IR group, which was comparable (Table 3). After 3 months of treatment, the mean change from baseline in resting HR was 1.1 ± 8.42 bpm in the ivabradine PR group vs. 0.0 ± 7.99 bpm in the ivabradine IR group (p = 0.5225). The least-square mean (standard error [SE]) change in HR from baseline to 3 months was 0.76 (1.188; 95% CI −1.59: 3.11) in the ivabradine PR vs. IR groups, which was well within the pre-specified margin of 6.5 bpm, confirming the non-inferiority of ivabradine PR (per-protocol analysis set). No age- or gender-specific effects were observed on actual or change from baseline to 3 months in resting HR due to the study treatment.
Holter Subgroup
There was no significant difference at the baseline or after 3 months between the groups in terms of resting HR, mean 24-h HR, mean HR (awake), or mean HR (asleep) (Table 4). Change from baseline (reduction in HR) to 3 months was comparable between the treatment groups for 24-h Holter ECG monitoring (least square mean [SE] −2.20 [2.410]; 95% CI −7.19:2.78; p = 0.3701), mean HR awake (least square mean [SE] −2.25 [2.319]; 95% CI −7.04:2.55; p = 0.3423), and mean HR asleep (least square mean [SE] −4.01 [2.696]; 95% CI −9.59:1.56; p = 0.1501).
Further, the HR (at baseline and 3 months, and change from baseline) was comparable within the treatment groups for 12-lead ECG vs. different parameters of Holter ECG. A stable HR was seen in both groups as assessed by inter-parameter comparison of mean 24-h Holter monitoring, mean awake, and mean asleep HR values within the treatment groups.
Safety
The safety profile of ivabradine PR was comparable to that of ivabradine IR.
A total of 39 TEAEs were reported in 29 (16.2%) patients within the study cohort (Table 5). In both the PR and IR groups, the majority of TEAEs were unrelated to the study drugs. In the PR group, two events of bradycardia in two (2.2%) patients, one event of sinus bradycardia in one (1.1%) patient, and one event of nausea in one (1.1%) patient were possibly related to the ivabradine PR treatment. In the IR group, one event of cardiac failure in one (1.1%) patient and two events of sinus bradycardia in two (2.2%) patients were possibly related, and one event of abdominal discomfort in one (1.1%) patient, one event of dizziness in one (1.1%) patient, and an event of dyspnea in one (1.1%) patient were probably related to the ivabradine IR treatment. The bradycardia events reported were comparable between groups. The drug was discontinued in two patients due to bradycardia and one patient due to nausea in the PR group; the drug was interrupted in one patient due to an inguinal hernia. In the ivabradine IR group, an event of cardiac failure led to the discontinuation of the drug in one patient.
Two SAEs were reported in two patients receiving ivabradine PR (gastritis and inguinal hernia); one SAE was moderate, while the other was severe. Neither was considered to be drug-related, and both were resolved. A total of five SAEs (cardiac failure, cardiac congestive failure, lower respiratory tract infection, dizziness, and dyspnea) were reported in three patients in the ivabradine IR group, of which two SAEs were unrelated, while one SAE in a patient was possibly and two SAEs in a patient were probably related to the ivabradine IR treatment. All five SAEs were severe in nature. The SAE of cardiac failure was fatal, while the remaining four SAEs were resolved.
One patient in the IR group was hospitalized due to worsening of HF.
No clinically significant worsening of visual symptoms was recorded during the study. Further, no clinically significant abnormalities were observed in any of the laboratory parameters in patients in either of the groups during the study.
The mean proportion of patients who complied with the study drug (as assessed by means of pill counts) was 98.61 ± 2.21% in the ivabradine PR group and 98.44 ± 2.05% in the ivabradine IR group.
Discussion
Our study provides evidence that the once-daily PR formulation of ivabradine is non-inferior to the ivabradine IR twice-daily regime, when added to background guideline-based medical treatment, in patients with stable CHF. The safety profile was comparable between the treatment groups. Thus, patients stabilized on an ivabradine twice-daily regimen of 5 mg or 7.5 mg can be shifted to an ivabradine once-daily regimen to effectively maintain HR, with potentially added convenience and treatment compliance.
Heart failure is a common, debilitating condition that is associated with a complex drug regime and comorbidities frequently requiring lifelong management. High medication burden is often observed in patients with HF, which may be attributed to multidrug treatment with multiple dosing regimens. This polypharmacy and high pill burden may be overwhelming for patients, resulting in medication noncompliance. While compliance can be a multifaceted process, dosing frequency plays a critical role in improving compliance, contributing to the effectiveness of the regimen [22, 24, 25]. Interestingly, a large meta-analysis concluded that a once-daily regimen may lead to significantly better compliance than twice-daily dosing [24, 26]. Bae and colleagues used evidence from administrative claims to assess medication compliance with antidiabetic, antihyperlipidemic, antiplatelet, or cardiac agent treatment in more than one million patients. They observed a significantly higher medication possession ratio (MPR) with a once-daily versus twice-daily regimen, with mean MPR of 0.66 and 0.57 (p < 0.01), respectively [27]. Moreover, clinical effectiveness with a once-daily regimen may be no lower than with multiple daily dosing [28, 29].
The 2017 guideline on the management of HF stipulates the role of ivabradine in chronic HF NYHA class II–IV with systolic dysfunction in patients in sinus rhythm and whose HR is ≥ 75 bpm, in combination with standard therapy including β-blockers or when β-blocker therapy is contraindicated or not tolerated [11, 30]. Ivabradine is also indicated for the symptomatic treatment of chronic stable angina pectoris in adults with CAD with normal sinus rhythm and HR ≥ 70 bpm who are unable to tolerate β-blockers or for whom β-blockers are contraindicated, or is indicated in combination with β-blockers in patients inadequately controlled with an optimal β-blocker dose [31].
In this PROFICIENT study, we assessed the non-inferiority of a new PR once-daily formulation of ivabradine compared with the conventional IR twice-daily dosage formulation with regard to the maintenance of stable HR for a period of 3 months in patients with stable CHF with systolic dysfunction. An earlier study had indicated the comparative bioavailability of ivabradine PR and ivabradine IR under fed conditions, which is the recommended dosing condition for ivabradine (results presented elsewhere).
A total of 180 patients were enrolled in the study. The demographic and baseline characteristics were comparable between the two treatment groups, with men constituting the major proportion of enrolled patients. All patients presented with stable HFrEF as a relevant medical condition. Approximately 79% of patients had ischemic conditions, while a small proportion of patients (32%) presented a non-ischemic condition.
The reduction in HR with ivabradine has been shown to improve clinical outcomes in patients with HF beyond the improvements observed with β-blockers [10, 13,14,15,16]. Ivabradine selectively and specifically inhibits the pacemaker If ionic current, which acts on the sinoatrial node, resulting in a reduction in HR. In our study, the primary efficacy analysis met the statistical criteria for non-inferiority of ivabradine PR relative to the ivabradine IR comparator, providing relatively robust evidence of a treatment effect on the primary endpoint of change in resting HR from baseline to the end of 3 months, as assessed by standard 12-lead ECG.
We also wanted to understand whether there was any sex- or gender-based impact on the change in HR from baseline to 3 months, which was found to be comparable between the groups. This data corroborated the published literature reporting that the effect of ivabradine was seen in patients regardless of the predefined subgroup [10, 32].
Evidence shows that HR variability as depicted by 24-h Holter monitoring, daytime HR, and nighttime HR may be associated with prognostic outcomes [33]. Kurtoglu et al. reported an improvement in the time domain indices of HR variability in patients with nonischemic dilated cardiomyopathy after 8 weeks of treatment with ivabradine. This improvement was sustained both when awake and while asleep. This was attributed to the prolonged diastole leading to the improved cardiac blood supply and LV filling, beneficial effects on LV remodeling and reduced sympathetic influence, and enhanced vagal tone, leading to an improvement in sympathovagal balance [34]. Hence, in the subpopulation with HF, we also assessed the baseline HR and the effect of ivabradine PR and IR on resting, 24-h, awake, and asleep HR. Our data indicate that the change in HR from baseline to 3 months, as assessed by the resting, 24-h, awake, and asleep HR, was comparable between the groups (statistically nonsignificant). Considering the possibility of variation in resting HR based on the time and condition of the recordings, an attempt was also made in this study to understand whether 24-h Holter parameters (awake, asleep, or 24-h average HR) responded similarly to resting HR. Stable HR was noted in both groups as assessed by mean 24-h Holter, mean awake, and mean asleep HR monitoring.
No new safety concerns were reported in the study. The overall rates of AEs were similar between the ivabradine PR and IR groups. None of the events in either treatment group were unexpected, and all the events were consistent with the published literature [10, 13,14,15,16, 32]. Further, the study medications did not worsen the clinical conditions associated with visual signs or symptoms during the study period. Both study drugs were well-tolerated. These results indicate that ivabradine PR has an acceptable safety profile, similar to ivabradine IR.
Ivabradine has been shown to substantially reduce the incidence of cardiovascular death or hospitalization for worsening HF compared with placebo in patients with HF and LV systolic dysfunction, already receiving guideline-based HF therapy [35]. In this study, hospitalization for worsening HF, all-cause mortality, and mortality due to CV was reported for only one patient in the IR group, though the same was not observed in any patient in the PR group.
The study has a few limitations. The Holter subgroup analysis was not statistically powered; hence the subgroup findings need to be substantiated in a larger group. Further, long-term studies in a larger population are needed to assess the effect of ivabradine PR formulation on HF outcomes, though the effect of ivabradine in terms of benefits such as decreasing hospitalization rates is well-documented [36]. Moreover, although no unwarranted safety concerns were observed in this study, longer follow-up is needed in a larger population to gain insights in the real-life clinical setting.
Conclusion
Ivabradine PR was found to be non-inferior to ivabradine IR in the management of patients with stable CHF with systolic dysfunction, as assessed by the change in HR from baseline to the end of 3 months. Ivabradine once daily (10 mg/15 mg) effectively maintained the HR in these patients who were shifted from a twice-daily regimen of 5 mg or 7.5 mg, which may ensure convenience and compliance with treatment. Ivabradine PR and IR exhibited comparable safety profiles and similar effects with respect to the maintenance of HR on resting, asleep, awake, and 24-h HR. No new safety concerns were reported. Overall, the results support the use of ivabradine once-daily PR as an alternative to the conventional twice-daily ivabradine IR formulation and could improve treatment compliance. Considering the alarming burden of HF and stable angina in India, clinicians must emphasize the importance of medication compliance with adequate treatment measures, to achieve better control of the disease.
References
Ziaeian B, Fonarow GC. Epidemiology and aetiology of heart failure. Nat Rev Cardiol. 2016;13(6):368–78.
Huffman MD, Prabhakaran D. Heart failure: epidemiology and prevention in India. Natl Med J India. 2010;23(5):283–8.
Mishra S, Mohan JC, Nair T, et al. Management protocols for chronic heart failure in India. Indian Heart J. 2018;70(1):105–27.
Ponikowski P, Voors AA, Anker SD, et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2016;37(27):2129–200 (published correction appears in Eur Heart J. 2016 Dec 30).
Seferovic PM, Ponikowski P, Anker SD, et al. Clinical practice update on heart failure 2019: pharmacotherapy, procedures, devices and patient management. An expert consensus meeting report of the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail. 2019;21(10):1169–86.
Hartmann C, Bosch NL, de Aragão ML, Tierie E, Zytinski L, Baena CP. The effect of ivabradine therapy on heart failure patients with reduced ejection fraction: a systematic review and meta-analysis. Int J Clin Pharm. 2018;40(6):1443–53.
Yancy CW, Jessup M, Bozkurt B, et al. 2013 ACCF/AHA guideline for the management of heart failure: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation. 2013;128(16):1810–52.
Correction to: 2016 ACC/AHA/HFSA Focused update on new pharmacological therapy for heart failure: an update of the 203 ACCF/AHA Guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. Circulation. 2016;134(13):e298.
Ouwerkerk W, Voors AA, Anker SD, et al. Determinants and clinical outcome of uptitration of ACE-inhibitors and beta-blockers in patients with heart failure: a prospective European study. Eur Heart J. 2017;38(24):1883–900.
Swedberg K, Komajda M, Böhm M, et al. Ivabradine and outcomes in chronic heart failure (SHIFT): a randomised placebo-controlled study. Lancet. 2010;376(9744):875–85 (published correction appears in Lancet. 2010 Dec 11;376(9757):1988. Lajnscak, M [corrected to Lainscak, M]; Rabanedo, I Roldan [corrected to Rabadán, I Roldan]; Leva, M [corrected to Ieva, M]).
Badu-Boateng C, Jennings R, Hammersley D. The therapeutic role of ivabradine in heart failure. Ther Adv Chronic Dis. 2018;9(11):199–207.
Kitai T, Tang WH. Pathophysiologic insights into heart rate reduction in heart failure: implications in the use of beta-blockers and ivabradine. Curr Treat Options Cardiovasc Med. 2016;18(2):13.
Swedberg K, Komajda M, Böhm M, et al. Effects on outcomes of heart rate reduction by ivabradine in patients with congestive heart failure: is there an influence of beta-blocker dose? findings from the SHIFT (Systolic Heart failure treatment with the I(f) inhibitor ivabradine Trial) study. J Am Coll Cardiol. 2012;59(22):1938–45.
Fox K, Ford I, Steg PG, et al. Heart rate as a prognostic risk factor in patients with coronary artery disease and left-ventricular systolic dysfunction (BEAUTIFUL): a subgroup analysis of a randomised controlled trial. Lancet. 2008;372(9641):817–21.
Fox K, Ford I, Steg PG, et al. Ivabradine for patients with stable coronary artery disease and left-ventricular systolic dysfunction (BEAUTIFUL): a randomised, double-blind, placebo-controlled trial. Lancet. 2008;372(9641):807–16.
Böhm M, Borer JS, Camm J, et al. Twenty-four-hour heart rate lowering with ivabradine in chronic heart failure: insights from the SHIFT Holter substudy. Eur J Heart Fail. 2015;17(5):518–26.
Yamanaka T, Onishi K, Tanabe M, et al. Force- and relaxation-frequency relations in patients with diastolic heart failure. Am Heart J. 2006;152(5):966.e1–7.
Tsutsui H, Momomura SI, Yamashina A, et al. Efficacy and safety of ivabradine in Japanese patients with chronic heart failure—J-SHIFT Study. Circ J. 2019;83(10):2049–60.
Esmaeili R, Matlabi M, Khajavi A, Aliasghari E, Sajjadi M. Factors Affecting Adherence to Antihypertensive Medication: results from a Rural Population Study in East of Iran. Glob J Health Sci. 2017;9(5):286–92.
Seth S, Ramakrishnan S, Parekh N, Karthikeyan G, Singh S, Sharma G. Heart failure guidelines for India: Update 2017. J Pract Cardiovasc Sci. 2017;3(3):133–8.
Wu JR, Moser DK. Medication adherence mediates the relationship between heart failure symptoms and cardiac event-free survival in patients with heart failure. J Cardiovasc Nurs. 2018;33(1):40–6.
Laliberté F, Bookhart BK, Nelson WW, et al. Impact of once-daily versus twice-daily dosing frequency on adherence to chronic medications among patients with venous thromboembolism. Patient. 2013;6(3):213–24.
Weeda ER, Coleman CI, McHorney CA, Crivera C, Schein JR, Sobieraj DM. Impact of once- or twice-daily dosing frequency on adherence to chronic cardiovascular disease medications: a meta-regression analysis. Int J Cardiol. 2016;216:104–9.
Coleman CI, Roberts MS, Sobieraj DM, Lee S, Alam T, Kaur R. Effect of dosing frequency on chronic cardiovascular disease medication adherence. Curr Med Res Opin. 2012;28(5):669–80.
Laliberté F, Nelson WW, Lefebvre P, Schein JR, Rondeau-Leclaire J, Duh MS. Impact of daily dosing frequency on adherence to chronic medications among nonvalvular atrial fibrillation patients. Adv Therapy. 2012;29(8):675–90.
Iskedjian M, Einarson TR, MacKeigan LD, et al. Relationship between daily dose frequency and adherence to antihypertensive pharmacotherapy: evidence from a meta-analysis. Clin Ther. 2002;24(2):302–16.
Bae JP, Dobesh PP, Klepser DG, et al. Adherence and dosing frequency of common medications for cardiovascular patients. Am J Manag Care. 2012;18(3):139–46.
Nakao A, Hisata K, Fujimori M, Matsunaga N, Komatsu M, Shimizu T. Amoxicillin effect on bacterial load in group A streptococcal pharyngitis: comparison of single and multiple daily dosage regimens. BMC Pediatrics. 2019;19(1):205.
Mavros MN, Polyzos KA, Rafailidis PI, Falagas ME. Once versus multiple daily dosing of aminoglycosides for patients with febrile neutropenia: a systematic review and meta-analysis. J Antimicrob Chemother. 2011;66(2):251–9.
Yancy CW, Jessup M, Bozkurt B, et al. 2017 ACC/AHA/HFSA focused update of the 2013 ACCF/AHA Guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. Circulation. 2017;136(6):e137–e161161.
Procorolan summary of product characteristics. https://www.servier.hk/sites/default/files/spc-pil/coralan_smpc.pdf. Accessed on 3 Aug 2020.
Tavazzi L, Swedberg K, Komajda M, et al. Efficacy and safety of ivabradine in chronic heart failure across the age spectrum: insights from the SHIFT study. Eur J Heart Fail. 2013;15(11):1296–303.
Johansen CD, Olsen RH, Pedersen LR, et al. Resting, night-time, and 24 h heart rate as markers of cardiovascular risk in middle-aged and elderly men and women with no apparent heart disease. Eur Heart J. 2013;34(23):1732–9.
Kurtoglu E, Balta S, Karakus Y, et al. Ivabradine improves heart rate variability in patients with nonischemic dilated cardiomyopathy. Arq Bras Cardiol. 2014;103(4):308–14.
Tardif JC, O'Meara E, Komajda M, et al. Effects of selective heart rate reduction with ivabradine on left ventricular remodelling and function: results from the SHIFT echocardiography substudy. Eur Heart J. 2011;32(20):2507–15.
Komajda M, Tavazzi L, Swedberg K, et al. Chronic exposure to ivabradine reduces readmissions in the vulnerable phase after hospitalization for worsening systolic heart failure: a post-hoc analysis of SHIFT. Eur J Heart Fail. 2016;18(9):1182–9.
Acknowledgements
This was a registration study for manufacturing and marketing approval permission. The authors would like to thank the participants and the research staff at the centers involved in the study.
PROFICIENT Trial Committee
Dr. Ambuj Roy (AIIMS, New Delhi), Dr. Aditya Kapoor (Sanjay Gandhi Post Graduate Institute of Medical Sciences, Lucknow), Dr. Balagopal Nair (Abbott), Dr. Deepak Chand Jangid (K.R. Hospital Mysore Medical College & Research Institute, Mysore), Dr. Dheeraj Kumar Gandotra (BLK Super Specialty Hospital, New Delhi), Dr. Jejoe Karan Kumar (Abbott), Dr. Leena Shah (KEM Hospital & Research Centre, Pune), Dr. Shivani Acharya (Abbott), Dr. Shubhangi Desai (Abbott), Dr. Subhash Gupta (Maharaja Agrasen Hospital, New Delhi), Dr. Sudhir Pawar (LTMMC & LTMGH, Mumbai), Suhas Talele (Abbott), Dr. Swati Biswas (Abbott), Dr. Tapan. Umesh (KEM Hospital & Research Centre, Pune).
The PROFICIENT Investigators
Ajay Mahajan (Department of Cardiology, LTMMC & LTMGH, Mumbai-400022, Maharashtra, India), B. B. Chanana (Department of Cardiology, Maharaja Agrasen Hospital, West Punjabi Bagh, New Delhi-110026, India), Dhiman Kahali (Department of Cardiology, B.M Birla Heart Research Center, 1/1, National Library Avenue, Kolkata -700027, West Bengal, India), Dinesha Basavanna (Department of Cardiology, K. R Hospital, Mysore Medical College & Research Institute, Irwin Road, Mysore-570001, Karnataka, India), Divya Prakash (Department of Cardiology, 2nd Floor, PMSSY Super Speciality Hospital, Victoria Hospital Campus, Bengaluru-560002, Karnataka, India), Manoj Chopda (Cardiology Department, Chopda Medicare & Research Centre Pvt. Ltd., 3/5, Patil lane no.1, Laxminagar, Near KBH Vidyalaya Canada Corner, Nashik-422005, Maharashtra, India), Milind Gadkari (Department of Cardiology, KEM Hospital & Research Centre, 5th floor, Diamond Jubilee Building, 489, Rasta Peth, Sardar Moodliar Road, Pune-411011, Maharashtra, India), Mukul Misra (Department of Cardiology, and Dean, Dr Ram Manohar Lohia Institute of Medical Sciences, Gomtinagar, Lucknow-226010, UP, India), Rajendra Kumar Premchand (Department of Cardiology Krishna Institute of Medical Sciences, 1-8-31/1, Minister Road, Secunderabad-500003, Telangana, India), Rajpal K. Abhaichand (Department of Cardiology, G. Kuppuswamy Naidu Memorial Hospital, Post Box No: 6327, Nethaji Road, Pappanaickenpalayam, Coimbatore-641037, Tamil Nadu, India), Rakesh Kumar Aggarwal (Deep Heart Center, Deep Hospital, 478 -L, Model Town, Ludhiana-141002, Punjab, India), Santanu Guha (Department of Cardiology, Kolkata Medical College and Hospital, MCH building 88, College Street, Kolkata-700073, West Bengal, India), G. Sengottuvelu (Department of Cardiology, Apollo Hospitals, 21 Greams Road, Chennai-600006, Tamil Nadu, India), Subhash Chandra (Department of Cardiology, BLK Super Speciality Hospital, OPD 1st Floor, Dr. B. L. Kapur Memorial Hospital Building No-5, Pusa Road, New Delhi-110005, India), Sudeep Kumar (Department of Cardiology, Sanjay Gandhi Post Graduate Institute of Medical Sciences (SGPGI), Raibareli Road, Lucknow-226014, UP, India), Tejas Patel (Department of Cardiology, Apex Heart Institute (A unit of TCVS PVT. Ltd.), Block G-K Mondeal Business Park, Near Gurudwara, S.G. Highway, Ahmadabad-380054. Gujarat, India), Uday Kumar Hosad (Department of Cardiology, Yashoda Hospital, Behind Hari hara Kalabavan, S P Road, Secunderabad- 500003, Telangana, India), Upendra Kaul (Department of Cardiology, Batra Heart Centre, Batra Hospital and Medical Research Centre, 1 Tughlakabad Institutional Area, Mehrauli Badarpur Road, New Delhi -110062, India), R. V. Vijay Bhaskar (Department of Cardiology, Gleneagles Global 6-1-1070/1 To 4, Lakdikapool, Hyderabad- 500004, Telangana, India).
Funding
This research and the journal’s Rapid Service Fee was funded by Abbott Healthcare Pvt. Ltd.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Medical Writing and Editorial Assistance
Dr. Shalini Nair, who is employed by Abbott, provided support on medical writing of this article.
Disclosures
Ajit Mullasari was the lead principal investigator in the study. All investigators received support from Abbott to conduct this study. Dr Mukul Misra is currently associated with Chandan Hospital (Director Cardiology), Lucknow, UP, India. Dr. R. V. Vijay Bhaskar is currently associated with KIMS Hospitals (Senior Consultant Interventional Cardiologist), Kondapur, Hyderabad, Telangana, India.
Compliance with Ethics Guidelines
The study protocol and amendments were approved by the respective institutional ethics committees of each site (full list of IRBs is included as supplementary material). The study was carried out per the protocol and in compliance with the International Council for Harmonisation Guideline for Good Clinical Practice, Declaration of Helsinki, Schedule Y (amended version, 2013) of Central Drugs Standard Control Organization, Ministry of Health and Family Welfare, Government of India, and ethical guidelines for biomedical research on human participants, Indian Council of Medical Research. Written informed consent was obtained from all patients before enrollment. Additional consent was obtained from patients who participated in the 24-h Holter ECG monitoring subgroup analysis. This study is registered at Clinical Trials Registry–India (CTRI/2018/04/013464 [trial registered prospectively on 24/04/2018]).
Data Availability
The data sets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
Digital Features
To view digital features for this article go to https://doi.org/10.6084/m9.figshare.12932993.
List of author present in PROFICIENT investigators are available in acknowledgment section.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Mullasari, A., The PROFICIENT investigators. Efficacy and Safety of Ivabradine Once-Daily Prolonged-Release versus Twice-Daily Immediate-Release Formulation in Patients with Stable Chronic Heart Failure with Systolic Dysfunction: A Randomized, Double-Blind, Phase 3 Non-Inferiority (PROFICIENT) Study. Cardiol Ther 9, 505–521 (2020). https://doi.org/10.1007/s40119-020-00200-8
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40119-020-00200-8