
Abstract
We report a rare adult case of mesoblastic nephroma with pulmonary metastasis. A 28-year-old man was referred to our hospital with a complaint of left flank pain and intermittent gross hematuria. A diagnosis of a left renal tumor was made after several radiographic examinations. But, a nodule was also detected in the lung by CT scan. We operated on him for left radical nephrectomy. On the basis of the histological analysis, we diagnosed it as adult mesoblastic nephroma (MN). Subsequently, he underwent follow-up at 25 months after the operation. Although there was no evidence of recurrence of the primary tumor lesion, the nodule in the left upper lobe was enlarged. Cutting needle core biopsy was therefore performed and the resultant data showed that the metastatic development of adult MN had occurred. We diagnosed the patient as having metastasis of cellular adult MN in the lung before the operation and postoperative metastatic progress. Local radiofrequency catheter ablation failed to treat the lung metastasis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Mesoblastic nephroma (MN) is a tumor originating from the undifferentiated mesenchymal tissues in the kidney and was first described by Bolande et al. [1]. MN is subdivided into “classical” and “cellular” forms on the basis of the degree of cellularity and mitotic activity: stromal cellularity is low for the classic variant but high for the cellular variant [2, 3]. The cellular variant may occur in adulthood and behave aggressively in contrast to the classical one. The cellular variant occurs mostly in infants and its occurrence in adults is extremely rare. Block et al. [4] described a case of a 32-year-old woman in 1973, which was the first reported case in adulthood. Furthermore, the adult cases with distant metastasis, even after the total removal of the primary tumor, are much rarer [5]. Jin et al. [6] reported the only case of cellular MN in adulthood, which recurred as a large metastatic lung mass 7 years after the total removal of the primary renal tumor and was diagnosed after left upper lobe lobectomy and lymph node dissection in the vicinity. Because of its rare metastasis to the lung, chest physicians are generally not familiar with its clinical features and treatment modalities. We report a case of cellular MN in adulthood, which progressed as a nodule in the lung within 2 years after the total removal of the primary renal tumor.
Case report
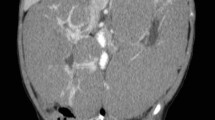
The patient was a 28-year-old man admitted to the urology department because of left flank pain and intermittent gross hematuria. On ultrasonography, a large mass in the left kidney was detected. Abdominopelvic computed tomography (CT) with intravenous and oral contrast media showed that the mass originated from the left kidney and had a maximum superoinferior diameter of 5 cm (Fig. 1a). Chest CT scan revealed a nodule in the left upper lobe of lung (Fig. 2a). There was no lymph nodule enlargement in the lungs. The patient underwent left radical nephrectomy. Histologically, the tumor was composed of both epithelial and mesenchymal components. Pathologic examination was in favor of MN, a cellular variant consisting of epithelial and stromal components. An extensive immunohistochemical analysis was carried out on the surgical specimen, which was highly positive for cytokeratins (CK) in the epithelial lining of the tubular and cystic structures and for vimentin in the fusocellular stroma of the tumor (Fig. 3).

CT of left renal tumor. The CT scan shows a well-circumscribed, heterogeneous, non-enhanced mass with dimensions 5.5 × 5.0 cm (a). There was no evidence of intra-abdominal tumor recurrence on abdominal CT scan after operation (b)

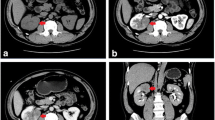
Chest CT scan reveals a nodule in the left upper lobe before operation (a), postoperation (b), and after radiofrequency catheter ablation (c)

Microscopic examination of renal tumor (×400). a H&E staining shows that the hypercellular areas are composed of uniform spindle cells carrying elongated nuclei. b CK staining was positive in the areas of epithelial components. c Vimentin staining was positive in the areas of stromal components
Because of the benign nature of the tumor, the patient did not receive chemotherapy but instead of being advised to undergo follow-up for the nodule in the lung. He underwent follow-up at 25 months after the operation. On abdominal CT scan, there was no evidence of intra-abdominal tumor recurrence (Fig. 1b). Brain CT did not show any abnormal finding. On chest CT scan, the nodule in the left upper lobe was enlarged (Fig. 2b). Then, cutting needle core biopsy was performed. Microscopically, the obtained specimen consisted of a monotonous population of interlacing bundles of spindle cells with long spindle-shaped nucleus and eosinophilic cytoplasm. By immunohistochemical analysis, the specimen was positive for vimentin and negative for CK, which suggested it was the metastasis of spindle cells in MN (Fig. 4). Therefore, we came to the conclusion that it was a metastasis of cellular adult MN in the lung before the operation and metastatic progress postoperatively. Then, radiofrequency catheter ablation was performed to treat the lung metastasis. After therapy for 8 months, chest CT scan showed a large mass in the left upper lobe. This suggested that radiofrequency catheter ablation failed to treat the lung metastasis in this case of cellular adult MN.

Microscopic examination of nodule in the left upper lobe. a H&E staining shows that the hypercellular areas are composed of uniform spindle cells carrying elongated nuclei with evenly distributed chromatins and inconspicuous nucleoli. b Vimentin staining was positive in the areas of stromal components
Discussion
MN is a distinctive tumor that is seen mostly in early infancy and occurs in two variants, i.e., classical and cellular (atypical). In most cases, there has been neither vascular nor lymphatic invasion, which is in accordance with the benign course of this neoplasm. MN rarely occurs in adulthood and it still is poorly characterized in this age group. Adult MN displays a distinctive morphologic spectrum that parallels that of its pediatric congener [7, 8]. The collecting duct differentiation expressed by most tubules and cysts of adult MN implies that the ureteral bud, which is the exclusive embryologic origin of the collecting duct, is an important element in the histogenesis of this rare but fascinating type of tumor [9]. Torres Gómez et al. [5] showed that epithelial elements with tubular conformation are surrounded by a spindle cell component, which is very useful in performing the differential diagnosis between this entity and others of greater clinical significance. Daniel et al. [7] reported a case with a complex pattern of antigenic expression not restricted to the collecting ducts, but including the glycoprotein CD24 and the neural cell adhesion molecule (NCAM). Trillo [10] reported the case of MN in a 41-year-old woman. The tumor was composed mainly of compact fibrocollagenous elements interspersed with areas containing immature tubules and occasionally glomeruloid structures. The stromal cells were composed of fibroblasts, myofibroblasts, and smooth muscle cells in various combinations. In our case, the spindle cells were distributed in the tumor. We used the CK and vimentin staining to identify the epithelial and stromal components. Cytokeratins (CKs, or following more recent nomenclature also simply called keratins) are intermediate filament-forming proteins that provide mechanical support and fulfill a variety of additional functions in epithelial cells. The specific nature of these heterodimers serves to distinguish the different epithelial cells in which they are expressed and has also become important in the classification of tumor cells. Vimentin is the major protein subunit of the 10-nm or intermediate filament proteins found in many kinds of mesenchymal cells, which provide structural support for the extensive microtentacles observed in detached tumor cells and a mechanism to promote successful metastatic spread. Vimentin is also found in many cell types in tissue culture, most notably fibroblasts, and in developing neuronal and astrocytic precursor cells in the central nervous system. The tumor was highly positive for CK in the epithelial lining of the tubular and cystic structures and for vimentin in the fusocellular stroma of the tumor, which implied that the tumor consisted of epithelial and stromal components.
MN is typically a benign tumor that can be treated successfully by complete excision. Rarely, however, cases with local recurrence or distant metastasis have been reported, particularly with the cellular variant of the tumor. Bell and Goodman [11] described the imaging appearances of an unusual, predominantly perinephric cystic congenital mesoblastic nephroma (CMN), with relative renal preservation but with retroperitoneal extension and bowel infiltration, which was complicated by hepatic metastases. Patel et al. [12] described a patient with isolated metastasis to the liver. Ali et al. [13] described a patient with CMN with metastasis to the brain. Vujanić et al. [14] highlighted a patient that was clinically and echographically free of hepatic or abdominal recurrences, and had good renal function as determined by a renogram and a normal serum creatinine, but had MN metastasis to the lungs and heart. Heidelberger et al. [15] reported a CMN metastasis to the brain; the appearance of the brain metastasis corresponded to that of the cellular nodule on histopathologic examination. Here, we report a rare adult case of MN with pulmonary metastasis. Although there was no evidence of recurrence of the primary tumor lesion, the metastatic nodule in the left upper lobe was enlarged. Microscopically, the obtained specimen consisted of spindle cells. By immunohistochemical analysis, the specimen was positive for vimentin and negative for CK, which suggested it was the metastasis of spindle cells in MN. We diagnosed the patient as having metastasis of cellular adult MN in the lung before the operation and postoperative metastatic progress. Then, radiofrequency catheter ablation was performed to treat the lung metastasis; however, a chest CT scan at 8 months after therapy suggested that radiofrequency catheter ablation had failed to treat the lung metastasis.
Although the histogenesis of MN remains obscure, its relationships to other pediatric neoplasms have been proposed. In 1998, Knezevich et al. [16] demonstrated that t(12;15)(p13;q25)-associated ETV6-NTRK3 gene fusions previously described in congenital fibrosarcoma were also present in cellular MN. The ETV6-NTRK3 fusion transcript was positive in partial cellular MNs and was all negative in four classical MNs. From these results, they assumed that classical and cellular MN had different genetic features and that cellular MN was histogenetically related to congenital fibrosarcoma. The biological consequence of this translocation is the expression of a chimeric tyrosine kinase with potent transforming activities in fibroblasts [16]. Watanabe et al. [17] demonstrated that the duplicated paternal IGF2 resulted in elevated IGF2 mRNA levels, and may provide CMN or its precursor cells with a proliferative advantage.
Sugimura et al. [18] identified a set of genes that distinguish MN from Wilms tumor (WT). Among this group of genes, topoisomerase II-alpha, which is highly expressed in WTs, is not overexpressed in MN. Witte et al. [19] presented evidence that an acidic fibroblast growth factor-like activity was detected in a primary MN. Taken together, atypical MN should be recognized as a potentially aggressive lesion separate from CMN.
Most of the patients with MN have a good prognosis if the tumor is completely excised. Because the number of reported cases is limited, the standard therapy for recurrent MN has not been established. Vujanić et al. [14] recommended that surgical resection of the recurrent lesion be performed if it is operable and that chemotherapy be considered only in unresectable cases because of its possible adverse effects. Recently, however, Loeb et al. [20] described three patients with recurrent cellular MN who showed complete responses to chemotherapy and suggested that chemotherapy be considered as a part of the therapy for recurrent or unresectable cellular MN. Furthermore, McCahon et al. [21] presented the possible relationship between the ETV6-NTRK3 fusion gene and the chemo-responsiveness of MN. Although there was no evidence of recurrence of the primary tumor lesion in our patient, the metastatic nodule in the left upper lobe was enlarged, which indicated the development of tumor. Even though the optimal strategies for recurrent MN remain unclear, in the future, it might be possible to tailor the treatment modalities according to the risk factors, with the expansion of experiences with MN.
References
Bolande RP, Brough AJ, Izant RJ Jr (1967) Congenital mesoblastic nephroma of infancy. A report of eight cases and the relationship to Wilms’ tumor. Pediatrics 40:272–278
Pettinato G, Manivel JC, Wick MR et al (1989) Classical and cellular (atypical) congenital mesoblastic nephroma: a clinicopathologic, ultrastructural, immunohistochemical, and flow cytometric study. Hum Pathol 20:682–690
Van Velden DJ, Schneider JW, Allen FJ (1990) A case of adult mesoblastic nephroma: ultrastructure and discussion of histogenesis. J Urol 143:1216–1219
Block NL, Grabstald HG, Melamed MR (1973) Congenital mesoblastic nephroma (leiomyomatous hamartoma): first adult case. J Urol 110:380–383
Torres Gómez FJ, Silva Abad A, Galán P (2007) Adult’s mesoblastic nephroma. Report of a case with aggressive course. Arch Esp Urol 60:72–75 (in Spanish)
Jin WM, Kil DK, Dong HS et al (2005) Pulmonary metastatic recurrence of mesoblastic nephroma in adulthood. Respir Med Extra 1:57–59
Daniel L, Lechevallier E, Bouvier C et al (2000) Adult mesoblastic nephroma. Pathol Res Pract 196(2):135–139
Truong LD, Williams R, Ngo T et al (1998) Adult mesoblastic nephroma: expansion of the morphologic spectrum and review of literature. Am J Surg Pathol 22:827–839
Yanai H, Ikeda A, Kadena H et al (2000) Adult mesoblastic nephroma with ciliated epithelium. A case report. Pathol Res Pract 196(4):265–268
Trillo AA (1990) Adult variant of congenital mesoblastic nephroma. Arch Pathol Lab Med 114:533–535
Bell MG, Goodman TR (2002) Perinephric cystic mesoblastic nephroma complicated by hepatic metastases: a case report. Pediatr Radiol 32:829–831
Patel Y, Mitchell CD, Hitchcock RJ (2003) Use of sarcoma-based chemotherapy in a case of congenital mesoblastic nephroma with liver metastases. Urology 61(6):1260
Ali AA, Finlay JL, Gerald WL et al (1994) Congenital mesoblastic nephroma with metastasis to the brain: a case report. Am J Pediatr Hematol Oncol 16:361–364
Vujanić GM, Delemarre JF, Moeslichan S et al (1993) Mesoblastic nephroma metastatic to the lungs and heart—another face of this peculiar lesion: case report and review of the literature. Pediatr Pathol 13:143–153
Heidelberger KP, Ritchey ML, Dauser RC et al (1993) Congenital mesoblastic nephroma metastatic to the brain. Cancer 72:2499–2502
Knezevich SR, Garnett MJ, Pysher TJ et al (1998) ETV6-NTRK3 gene fusions and trisomy 11 establish a histogenetic link between mesoblastic nephroma and congenital fibrosarcoma. Cancer Res 58:5046–5048
Watanabe N, Haruta M, Soejima H (2007) Duplication of the paternal IGF2 allele in trisomy 11 and elevated expression levels of IGF2 mRNA in congenital mesoblastic nephroma of the cellular or mixed type. Genes Chromosom Cancer 46:929–935
Sugimura J, Yang XJ, Tretiakova MS et al (2004) Gene expression profiling of mesoblastic nephroma and Wilms tumors—comparison and clinical implications. Urology 64:362–368
Witte DP, Nagasaki T, Stambrook P et al (1989) Identification of an acidic fibroblast growth factor-like activity in a mesoblastic nephroma. Lab Invest 60:353–359
Loeb DM, Hill DA, Dome JS (2002) Complete response of recurrent cellular congenital mesoblastic nephroma to chemotherapy. J Pediatr Hematol Oncol 24:478–481
McCahon E, Sorensen PH, Davis JH et al (2003) Non-resectable congenital tumors with the ETV6-NTRK3 gene fusion are highly responsive to chemotherapy. Med Pediatr Oncol 40:288–292
Acknowledgments
This work was supported by the National Natural Science Foundation of China (project 81000919).
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding authors
Additional information
C. Chen and Y.-H. Zhu contributed equally to this article.
About this article
Cite this article
Chen, C., Zhu, YH., Ni, CJ. et al. Development of pulmonary metastasis after removal of the primary tumor: a case report of an adult mesoblastic nephroma and review of the literature. Int Canc Conf J 1, 190–194 (2012). https://doi.org/10.1007/s13691-012-0037-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13691-012-0037-y