
Abstract
Endocannabinoid system is related with various physiological and cognitive processes including fertility, pregnancy, during pre- and postnatal development, pain-sensation, mood, appetite, and memory. In the latest decades, an important milestone concerning the endocannabinoid system was the discovery of the existence of the cannabinoid receptors CB1 and CB2. Anandamide was the first reported endogenous metabolite, which adjusted the release of some neurotransmitters through binding to the CB1 or CB2 receptors. Then a series of cannabinomimetric lipids were extracted from marine organisms, which possessed similar structure with anandamide. This review will provide a short account about cannabinomimetric lipids for their extraction and synthesis.
Similar content being viewed by others

1 Introduction
In the past decades, pharmacologists devoted more interest to the study of endocannabinoid system due to its relation with various physiological and cognitive processes including fertility, pregnancy, during pre- and postnatal development, pain-sensation, mood, appetite, and memory [1]. Originally, the endocannabinoid system was discovered while scientists tried to understand the physical and psychological effects of cannabis, thereby named it the endocannabinoid system for this reason. An important milestone concerning the endocannabinoid system was the discovery of the existence of the cannabinoid receptors (CB1 and CB2) in central and peripheral mammalian tissues [2,3,4,5]. Both receptors CB1 and CB2 belong to the large family of G-protein-coupled receptors (GPCR). CB1 receptor exhibits a widespread distribution in the mammalian brain and are responsible for the psychological and anticonvulsive effects produced by marijuana [2,3,4,5], while CB2 receptor is most abundant in the immune and hematopoietic system and is involved in the anti-inflammatory and possibly other therapeutic effects of cannabis [4, 5]. The discovery of cannabinoid receptors (CB1 and CB2) has launched the quest for endogenous ligands of these receptors. Based on the assumption that the endogenous cannabinoid ligand was a lipid soluble compound, a lipid derivative was first isolated from chloroform–methanol extracts of porcine brain and christened anandamide by Mechoulam et al. in 1992 (Fig. 1) [6]. This endogenous metabolite bound to both CB1 and CB2 receptors and was found in nearly all tissues in a wide range of animals [7]. Then a series of alkyl amides were extracted from marine organisms, which resembled structurally some aspects of anandamide and had been termed cannabimimetic lipids. In the biological activity tests, they showed the ability to bind and activate at least one cannabinoid receptor [8]. This review will provide a short account of cannabinomimetric lipids for their natural extract and artificial synthesis.

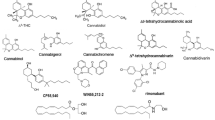
The structure of anandamide
2 Extraction and Biological Activities (Fig. 2)
2.1 Grenadamide
Grenadamide was isolated from the organic extract of a Grenada collection of the marine cyanobacterium Lyngbya majuscula by Gerwick et al. in 1998. It exhibited brine shrimp toxicity (LD50 = 5 μg/mL) and modest cannabinoid receptor binding activity (K i = 4.7 μM) [9]. Gerwick et al. verified the structure and the relative stereochemistry of grenadamide, which was a trans-cyclopropyl-containing fatty acid-derived metabolite.


The structures of cannabinomimetric lipids
2.2 Mooreamide A
Mooreamide A was extracted from cyanobacterium Moorea bouillonii by Gerwick et al. from Papua New Guinea, and showed strong and selective affinity to CB1 ligand [10].
2.3 Serinolamides
Serinolamide A was isolated from marine cyanobacteria Lyngbya majuscule collected in Papua New Guineal. It displayed a moderate agonist effect and selectivity for the CB1 cannabinoid receptor [11]. Serinolamide B, a closely related analogue of serinolamide A, was isolated from a Lyngbya sample from the piti Bomb Holes in Guam by Luesch et al. [12]. It showed moderate affinities to both CB1 and CB2, while exhibited a higher selectivity for CB2 (K i = 5.2 μm) over CB1 (K i = 16.4 μm)
2.4 Semiplenamides
Semiplenamides A to G were isolated from the marine cyanobacterium Lyngbya semiplena collected from Papua New Guinea by Gerwick et al. [13]. In the test of their affinity to cannabinoid receptors of the rat brain membranes, only semiplenamides A, B and G worked. In the test of fatty acid amide hydrolase (FAAH), the semiplenamides A to G were not found appreciable inhibitory effect.
2.5 Malyngamides
Malyngamides include over 30 members, characterized by different N-substitution groups of amides. They were isolated from Marine cyanobacterium Lyngbya majuscule, and showed a wide range of biological activities, such as antifeedant activity, ichthyotoxicity, toxicity to other marine animals, cytotoxicity to cancer cells, anti-HIV, anti-leukemic, and anti-tumor activity [14,15,16]. The finding that malyngamide B possesses cannabimimetic properties provides new insight into the biological activities of malyngamides. The extraction information of malyngamides was illustrated in Table 1.
Hermitamides resemble the malyngamide-type compound in structure and were still isolated from the marine marine cyanobacterium L. majuscula of other species of Gracilaria [17,18,19]. Hermitamides were evaluated for their biological activity in several systems. Hermitamides A (1) and B (2) showed LD50 values of 5 μM and 18 μM respectively in the brine shrimp (Artemia salina) toxicity assay, and showed IC50 values of 2.2 μM and 5.5 μM respectively to Neuro-2a neuroblastoma cells in tissue culture.
3 Synthesis of Cannabinomimetric Lipids
3.1 Synthesis of Grenadamide
In 2004, Baird and co-workers reported the synthesis of grenadamide and confirmed its absolute stereochemistry (Scheme 1) [9]. The synthesis started from the aldehyde 1, which was converted to olefin 2 through Wittig reaction the following ester hydrolysis. Then removal of the double bond gave 3. Oxidation of alcohol 3 got aldehyde 4 and epimerisation of 4 using sodium methoxide in methanol afforded the epimer 5. Then 5 underwent HWE reaction with ethoxycarbonyl triphenylphosphosphorane to give the ester 6, which was removed the double bond with di-potassium azodicarboxylate and hydrolysed with KOH to afford acid 7. The compound 7 was converted into the corresponding chloride, then treated with 2-phenylethylamine to give the amide 8, which had an equal and opposite absolute rotation compared with natural grenadamide. So the synthetic sample was the enantiomer of the natural product grenadamide.

Synthesis of grenadamide reported by Baird [9]
One year later, Bull and co-workers reported an asymmetric synthesis of grenadamide in 6 steps using (R)-5,5-dimethyl-oxa-zolidin-2-one as a chiral auxiliary (Scheme 2) [46]. The starting material 9 as a chiral auxiliary was acetylated with chloroacetyl chloride to give 10. Then treatment of 10 with 9-BBN–OTf and i-Pr2NEt and following reaction with α,β-unsaturated aldehyde afforded syn-aldol product 11 in 92% de. Cyclopropanation of 11 with Et2Zn and CH2I2 afforded 12 with high stereoselectivity. Then replacing the oxazolidin-2-one fragment with phenylethylamine gave 13, which was treated with SmI2 resulted in clean elimination reactions to afford (E)-α,β-unsaturated amide. Finally reduction of the double bond with NaBH4 and CoCl2 afford grenadamide.

Synthesis of grenadamide reported by Bull [46]
In the same year, Taylor and co-workers reported the total synthesis of both (+)-grenadamide and (−)-grenadamide by a racemic route (Scheme 3) [47]. Wittig reaction of 4-methoxycinnamaldehyde 15 and octyltriphenylphosphonium iodide resulted the 1,3-diene 16. Then photolysis of 1,3-diene 16 gave the 1,2-dioxine 17, which reacted with tert-butyl ester ylide to afford cyclopropane 18. Then hydrolysis of tert-butyl ester group with formic acid gave acid 19, which was subsequently decarboxylated using the Barton protocol to afford 20. Baeyer–Villiger oxidation of 20 using m-CPBA proceeded with excellent selectivity to give phenol ester 21, which was subsequently hydrolyzed to give cyclopropyl fatty acid 22. Arndt-Eistert homologation/amidation of acid 22 afforded racemic grenadamide. To the further study, the author obtained the two enantiomers of grenadamide. Fatty acid 22 was coupled with Evans’ auxiliary and chromatographically separating the diastereomers 23 and 24, which were removed of the auxiliary to give (−)-22 and (+)-22 respectively. Then the enantiomerically pure fatty acids were subjected to the Arndt-Eistert protocol to give (−)-grenadamide and (+)-grenadamide.

Synthesis of grenadamide reported by Taylor [47]
In 2007, Piva and co-workers reported the synthesis of racemic grenadamide through a sequential cross-metathesis/Simmons–Smith cyclopropanation (Scheme 4) [48]. Cross-metathesis of 26 with 1-nonene 25 catalyzed by Grubbs type catalyst 27 delivered 28 as mixture of E and Z isomers. Then cyclopropanation of the mixture of 28 afforded grenadamide.

Synthesis of grenadamide reported by Piva [48]
In 2010, Boysen and co-workers reported an asymmetric synthesis of (+)-grenadamide, an enantiomer of the natural product (−)-grenadamide (Scheme 5) [49]. The cyclopropyl carboxylic ester 29 was transformed into the corresponding aldehyde 30 by reduction with lithium aluminium hydride to alcohol, followed by Swern oxidation. Then aldehyde 30 underwent Wittig olefination to give α,β-unsaturated ester 31. Reduction of 31 and followed by hydrolysis afforded acid 32, which was coupled with phenethylamine gave (+)-grenadamide.

Synthesis of grenadamide reported by Boysen [49]
3.2 The Synthesis of Serinolamide A
In 2013, our group reported the first total synthesis of (+)-serinolamide A in nine steps from l-serine with 30% overall yield (Scheme 6) [50]. The synthesis of (+)-serinolamide A was divided into two parts, the fatty acid 35 and the derivatives of serinol 40 and 41. The coupling of 1-bromotridecane and pent-4-yn-1-ol using excess n-BuLi in HMPA afforded 34. Then reduction of 34 with LiAlH4 afforded alkene and following oxidation of hydroxyl with PDC gave fatty acid 35. The other key part was synthesized from l-serine. Esterification of 36, followed by protection of the amino group afforded 37. Methylation of 37 with iodomethane and then reduction of ether group with NaBH4 gave 38. Protection of the hydroxyl group with TBDMSCl, subsequent N-methyl using MeI and NaH afforded 39. Deprotection of Boc group gave the mixture of 40 and 41, which without purification and separation condensed with 35 to afford the corresponding products 42 and serinolamide A. Then 42 was treated with TBAF to convert into serinoamide A as well.

Synthesis of (+)-serinolamide A reported by Wang [50]
Later, Pandey and co-workers reported the synthesis of (+)-serinolamide A in 2015 from the starting material of butadiene monoepoxide in five steps with 51% yield (Scheme 7) [51]. The compound 43 under the Trost’s DyKAT conditions reacted with phthalimide furnished asymmetric allylic alkylation derivative pthaloyl alcohol 44 in high regio- and enantioselective. Etherification of 44 with MeI in presence of NaH furnished 45. Cleavage of phthalimide with hydrazine and then protection of the primary amine with (Boc)2O gave 46. Then oxidative cleavage of terminal double bond gave the aldehyde, which was under reduction conditions to afford the serino derivative 41. The other part fatty acid 3 was synthesized from pentadec-1-ene 47 and 4-pentenoic acid 48 undertaken RCM reaction. The fatty acid 35 was then condensed with serino derivative 41 to afford serinoamide A.

Synthesis of (+)-serinolamide A reported by Pandey [51]
3.3 Synthesis of Semiplenamides
In 2005, Bull and co-workers developed an efficient method for the synthesis of (E)-α,β-unsaturated amide and applied the methodology for the synthesis of semiplenamide C (Scheme 8) [52]. l-Alanine methyl ester 49 was chosen as the starting material. Reduction of 49 with LiAlH4 and then protection with diethyl carbonate afford 50. Subsequent treatment of 50 with n-BuLi and propionyl chloride gave 51. Then pretreatment of 51 with 9-BBN-OTf and i-Pr2NEt underwent an aldol reaction with tetradecanal to give 52 in > 95% de. Finally, deprotection of 52 with KOtBu afforded semiplenamide C.

Synthesis of semiplenamide C reported by Bull [52]
In 2009, Das and co-workers reported the synthesis of semiplenamides C and E through the Baylis–Hillman adducts [53]. (Scheme 9) The Baylis–Hillman adducts 53 and 54 were treated with PPh3/CBr4 afforded the corresponding allyl bromides 55 and 56, which were subsequently treated with Zn and CH3COOH to give 57, 58 respectively. The esters 57 and 58 were then hydrolyzed with KOH to give the corresponding acids 59 and 60, which were condensed with (S)-alaninol to form semiplenamide C (61) and 62 respectively. Compound 62 was further acetylated with acetic anhydride to furnish semiplenamide E.

Synthesis of semiplenamide C reported by Das [53]
3.4 Synthesis of Malyngamides
In 2006, Piva and co-workers reported both the racemic synthesis and a formal enantioselective synthesis of hermitamides A and B (Scheme 10) [54]. Homoallylic ether 64 was prepared through the Grignard reaction from the octanal 63 and allyl magnesium bromide and the following protection of the hydroxyl group by MeI in the presence of NaH. Then 64 and butenoic acid proceeded RCM reaction to afford 65 with the E/Z ratio of 95/5. Finally, racemic hermitamide A and B were synthesized through the condensation of 65 with the phenethylamine and 3-indolyl-ethylamine respectively. For the further study, the author developed an enantioselective methodology for the synthesis of 68, which was obtained through the reaction of (+)-camphor homoallylic alcohol 66 and octanal 67 catalyzed by CAS with the ee value of 85%.

Synthesis of hermitamides A and B reported by Piva [54]
In 2009, Frost and co-workers reported an enantioselective synthesis of hermitamides A and B from the starting material 1-nonene 25 (Scheme 11) [55]. Epoxidation of 25 by m-CPBA afforded the racemic epoxide 69. Then catalyzed by cobalt (II) complex 76, rac-69 underwent a hydrolytic kinetic resolution process to afford the chiral epoxide (S)-69 in 96% ee. Subsequently, homopropargylic alcohol 70 was obtained when the addition of lithium acetylide complexed with EDA. After methylated the hydroxyl group, the compound 71 was converted to the chiral alkenylpinacol boronic ester 72 with the addition of catalytic Schwartz reagent and anhydrous triethylamine. However, 72 was failed to react with both phenylethylamine and tryptamine acrylamide under a range of conditions. Thus pinacolboronic ester 72 was converted to chiral alkenyltrifluoroborate salt 73. Finally, alkenyltrifluoroborate salt 73 was reacted with phenylethylacrylamide 74 or 75 in the presence of [Rh(cod)(OH)]2 and cyclooctadiene as the ligand to afford hermitamides A and B respectively.

Synthesis of hermitamides A and B reported by Frost [55]
In 2011, Paige and co-workers reported an asymmetric synthesis of hermitamides A and B (Scheme 12) [56]. Asymmetric allylation of octanal with allyltributylstannane mediated by a titanium-binol complex gave homoallylic alcohol followed by the methylation with MeI to afford ether 77. Oxidative cleavage of the terminal double bond yielded aldehyde 78, which was reacted with vinylmagnesiun bromide to afford allylic alcohol 79. The compound 79 was then generated Johnson-Claisen rearrangement with the addition of trimethylorthoacetate in the presence of catalytic amount of propionic acid to afford methyl ester of lyngbic acid followed by the saponification with lithium hydroxide to give 65. Then acid 65 was coupled with phenethylamine or tryptamine to afford hermitamides A and B respectively.

Synthesis of hermitamides A and B reported by Paige [56]
In 2014, Narender and co-workers reported a concise total synthesis of hermitamides A and B in a high enantioselectivity (Scheme 13) [57]. The synthesis was commenced from octanal 63. Vinylation with vinyl magnesium bromide gave allylic alcohol 80, which was oxidated by IBX to afford enone 81. Then asymmetric reduction of ketone 81 catalyzed by CBS gave the chiral allylic alcohol with high enantioselectivity, which was treated with Meerwein’s reagent to afford the methylation product (S)-82. Subsequently, hydroboration-oxidation of 82 afforded the primary alcohol 83. Then 83 with 1-phenyltetrazole-5-thiol (84) underwent Mitsunobu reaction to give aryl sulfide 85, which was further oxidated by ammonium molybdate and hydrogen peroxide to give sulfone 86. Coupling of alkyl sulfone 86 with aldehyde 87 via Julia-Lythgoe olefin provided the corresponding olefin 88 with E-geometry exclusively. Then removal of benzyl obtained the primary alcohol following oxidation of the hydroxyl group to furnish lyngbic acid 65. Acid 65 was coupled with 2-phenylethyl amine or 3-indolylethylamine to provide hermitamides A and B, respectively.

Synthesis of hermitamides A and B reported by Narender [57]
In 2006, Cao and co-workers reported the synthesis of serinol-malyngamide for the first time (Scheme 14) [58]. The molecular was divided into two parts, the fatty acid 97 and the derivative of serinol 100. The stereoselective synthesis of fatty acid 97 had been reported previous [59], which was starting from 1-tetradecanol. Oxidation of 89 afforded the aldehyde 90, which further reacted with allyltributyltin catalyzed by bis-(R)-Ti(IV) oxide (91) to afford allyl alcohol 92. Methylation of hydroxyl group and followed by oxidation of terminal olefin afforded aldehyde, subsequently reacted with PPh3/CBr4 to produce dibromide 93. Then alkyne 94 was obtained after the addition of n-BuLi. Alkyne coupled with 95 gave 96, reduction of which obtained olefin with E-configuration. Sequent deprotection of THP group and oxidation of the obtained primary alcohol afforded fatty acid part 97. The other part of the derivative of serinol 100 would be synthesized from chiral starting material d-serine by the method of Meyers et al. Finally the acid 97 and the derivative of serinol 100 were condensed under the DCC conditions to afford serinol-malyngamide.

Synthesis of serinol-malyngamide reported by Cao [58]
In 2007, Isobe and co-workers reported the synthesis of malyngamide X [60], which possessed an unusual tripeptide portion connecting to a methoxylated fatty acyl chain (Scheme 15). Malyngamide X still was divided into two parts tripeptide portion 108 and fatty acid portion 65. The synthesis of tripeptide portion was started from commercially available N-Boc-l-valine (101), which was coupled with Meldrum’s acid (102) to afford pyrrolidone derivative 103. Then sequent Mitsunobu reaction, deprotection of Boc group and following N-propionylation occurred, N-propionyl pyrrolidone 104 was obtained. Subsequently, 104 was then coupled with N-Boc-l-alaninal (105) assisted by n-BuBOTf to afford the pyrrolidone derivative 106. Then the coupling of 106 and Boc-N-Me-l-alanine gave the desired tripeptide segment 108. The other part was started from R-glycidyl tosylate (109). Thus, Grignard reaction of 109 with C6H13MgBr gave the desired alcohol 110. Then conversion of alcohol 110 to epoxynonane (+)-69 and the following coupling with acetylide 111 gave homopropargylic alcohol 112. Methylation of alcohol and the subsequent reduction of acetylene provided 113 with E-geometry. Cleavage of THP protecting group and the following oxidation of hydroxyl group gave fatty acid 65. Finally, coupling of lyngbic acid segment 65 with amine 108 gave malyngamide X.

Synthesis of malyngamide X reported by Isobe [60]
In 2006, Cao and co-workers reported the total synthesis malyngamide U [61] and revised its correct absolute configuration. The later year, they reported an improved asymmetric synthesis of malyngamide U [62] from the same material and similar methodology (Scheme 16). The fatty acid 114 was synthesized as the same way illustrated in Scheme 13, and the starting material was hexanal instead of tetradecanal. Then amidation of acid 114 with ethanolamine and oxidation of the obtained primary alcohol provided amido-aldehyde 115. The other part started from (R)-(−)-carvone 116, which was indicated to convert to 120 for the further aldol reaction with amido-aldehyde 115. Epoxidation of (R)-(−)-carvone, reduction of ketone moiety under Luche’s conditions and following protection of the hydroxyl group with p-methoxybenzyl (PMB) generated 117. Then oxidation of the terminal double bond to ketone and further Baeyer–Villiger rearrangement of ketone with m-CPBA gave the corresponding acetate. Removal of acetyl group afforded the secondary alcohol 118. Subsequently the alcohol 118 was protected using allyl bromide and removal of the protecting group PMB with DDQ gave 119. Then the oxidation of the resulted alcohol got ketone by IBX and the reduction of the epoxide on the work of Adams provide the key intermediate 120. Aldol condensation of 120 with amido-aldehyde 115 afforded two epimers 121 and 122. The configuration of 122 was in accordance with malyngamide U. Thus methylation of 122 with MeI gave 123, which was also obtained by Mitsunobu reaction of 121. Finally, removal of the allyl protecting group completed the synthesis of malyngamide U.

Synthesis of malyngamide U reported by Cao [62]
In 2009, Cao and co-workers reported a convergent route for the total synthesis of malyngamides O, P, Q, and R (Scheme 17) [63]. Preparation of key intermediate 131 began with ethyl 4-chloro-3-oxobutanoate 124. Azidation of 124 afforded azide 125, which was subsequently hydrogenated by H2 in the presence of di-tert-butyl dicarbonate gave the Boc-protected amine 126. Reduction of both keto and ester carbonyl groups in ester 126 with diisobutylaluminum hydride (DIBAL-H) afforded the corresponding diol, followed by monoprotection of the primary hydroxy group with tert-butyldiphenylsilyl chloride (TBDPSCl) to give the corresponding silyl ether 127. Then oxidation of secondary alcohol 127 with 2-iodoxybenzoic acid (IBX) provided the corresponding ketone 128, which was subjected directly to Wittig olefination with chloromethyltriphenylphosphonium iodide (129) to give the vinyl chloride as a mixture of Z- and E-isomers (Z:E = 3:1). The Z-configuration of the vinyl chloride was consistent with that in natural malyngamides O and P. Then N-methylation of 130 provided the key vinyl chloride 131. Thus, deprotection of the TBDMS group of 131 with TBAF, followed by oxidation of alcohol with IBX afforded aldehyde 132. Then aldehyde 132 reacted with the enolate derived from methyl acetate in THF to give 133. Racemic alcohol 133 was immediately submitted to deprotection of the Boc group to generate the corresponding amine, which was directly condensed with the carboxylic acid 65 to afford amide 134. Finally, oxidation of 134 with Dess-Martin periodinane gave malyngamide P. Deprotionation and the following methylation provided malyngamide O. Then the authors continued to synthesize malyngamides Q and R, bearing the more challenging structure. The acetamide 138 bearing the pyrrolidone ring was prepared from l-serine 135. Protection of the amino group and hydroxy group in l-serine with Boc2O and TBSCl respectively provided acid 136. Then condensation reaction of 136 with Meldrum’s acid, followed by treatment with MeOH furnished the pyrrodidone intermediate, which was subjected to a Mitsunobu reaction to give O-methyl pyrrolidone derivative 137. Removal of the Boc group with TFA provided pure amine, which was further protected by acetyl chloride provided N-acetyl pyrrolidone (S)-138. However, aldol reaction products of 138 and 132 could be achieved, the further condensation reaction with acid 65 was failed with little desired compound. Thus another strategy was adopted that was amidation of 130 and 131 first, and then conducted the aldol reaction with (S)-138 in the second step. Removal of the Boc groups in 130 and 131, followed by amidation with acid 65 produced amides 141 and 142, respectively. Then deprotection of the TBDMS groups in 141 and 142 with TBAF gave the corresponding alcohols 143 and 144 respectively. N-Protection with (Boc)2O affored 145. Oxidation of alcohol 144 and 145, followed by condensation precursor enolate of pyrrolidone 138 afforded a diastereomeric mixture of alcohols 146 and 147 respectively. Oxidation of 146 and the following methylation of the enol afforded Malyngamide R. Using the similar route for the preparation of malyngamide R, enol methylation and subsequent removal of the Boc group gave malyngamide Q.


Synthesis of malyngamide O, P, Q and R reported by Cao [63]
In 2010, Cao and co-workers reported the stereoselective synthesis of malyngamide M [64], which was still divided into two parts (Scheme 18). The lyngbic acid part 65 was achieved through the common methodology reported by their group (Scheme 13). The other amine part 152 possessing a vinylic chloride moiety was synthesized from o-cresol 148. Friedel–Crafts acetylation of 148 with chloroacetonitrile and the following substitution reaction with sodium azide gave phenol 149. Then protection of phenol with methoxymethyl chloride and subsequent hydrogenation of azide group by 10% Pd/C in situ protection with di-tert-butyl dicarbonate afforded 150. Then 150 underwent Wittig olefination with chloromethyl triphenylphosphonium iodide to provided 151 with Z geometry. The amine part 152 was obtained through the sequent N-methylation and the simultaneous removal of both the MOM and Boc groups. Finally the coupling of acid part 65 and amine part 152 only afforded the isomalyngamide M with the Z-vinyl chloride in the structure, which was exposed to UV-light (λ > 300 nm) in the presence of benzophenone to afford malyngamide M (E-vinyl chloride).

Synthesis of malyngamide M reported by Cao [64]
In 2011, Cao and co-workers reported the total synthesis of malyngamide W and confirmed the absolute configuration of malyngamide W (Scheme 19) [65]. α,β-Unsaturated cyclohexanone 155 was a key intermediate towards the synthesis, which was synthesized from (R)-(−)-carvone 116. Epoxidation of 116 using hydrogen peroxide provided epoxy ketone 153. Then the epoxide was opened and led to the 3a-hydroxyl ketone. Subsequently, the hydroxy group was further protected by tert-butyldimethylsilyl chloride to provide its TBS-ether 154. Ozonolysis of 154 and subsequent treatment with copper (II) acetate monohydrate/iron sulfate heptahydrate furnished the enone 155. Iodination of 155 obtained α-indo-α,β-unsaturated cyclohexone 156, which underwent Luche’s reduction and subsequent protected by benzyl bromide to afford the key iodide 157. The other part was synthesized from 158, which was oxidized by IBX to provide the aldehyde 159. Then 157 and 159 underwent Nozaki–Hiyama–Kishi coupling reaction to afford 160, which was removed the N-protection group and coupled with lyngbic acid 65 to afford amide 161. Oxidation of 161 with Dess–Martin periodinane afforded 162. Reduction of 162 by Corey-Bakshi-Shibata oxazaborolidine (CBS catalyst) and methylation of the hydroxyl group afforded 163. Then removal of the O-Bn protecting group with DDQ and subsequent oxidation of the alcohol with DMP afford amide 164. Finally, removal of the TBS moiety gave malyngamide W.

Synthesis of malyngamide W reported by Cao [65]
In 2011, Cao group reported the total synthesis of malyngamides K (Scheme 20), L, and 5″-epi-C and confirmed absolute configuration of malyngamide L [66]. For the synthesis of malyngamide K, boronic acid part 167 began from 2-cyclohexen-1-one (165). Thus, bromination of enone 165 with bromine generated the bromoenone, followed by protection of the carbonyl group with ethylene glycol to afford the ketal 166. Ketal 166 was easily transformed to boronic acid 167 by treatment with trimethyl borate in the presence of n-butyllithium, and subsequent treatment with hydrogen chloride. The other part amides 172 and 174 began with ethyl propiolate 168, which was converted to ester 169 in the presence of n-tetrabutylammonium iodide. Then reduction of ester gave the intermediate alcohol 170 as a mixture of E- and Z-isomers (E:Z = 1.1:1). The E-isomer could be converted to the desired Z-isomer by irradiation with UV light (> 350 nm) in DCM. Thus, configuration of Z-170 was consistent with that in natural malyngamides K, L, and epi-C. Then protection of the hydroxyl group with p-toluenesulfonyl group afforded the tosylate 171, which was transformed into the intermediates 172 and 174, respectively. The synthesis of amide 172 was also achieved by treatment of tosylate 171 with an excess of aqueous methylamine and followed by amidation with acid 65. Azidation of 171 and reduction of the obtained azide 173, followed condensation with the acid 65 afforded 174. Then malyngamides K was achieved by the Suzuki coupling reaction of 174 and 167.

Synthesis of malyngamide K reported by Cao [66]
Then the authors completed the more complex malyngamides L, and 5″-epi-C (Scheme 21) [66]. For the synthesis of malyngamides 5″-epi-C, the key was the synthesis of boronic acid part 182. Oxidation of 175 using nitrosobenzene gave the corresponding hydroxylamine, which was reduced to afford a single alcohol under Luche conditions, followed by reductive cleavage of the N-O bond to afford diol 176. Then deketalization and elimination of diol 176 with hydrogen chloride in THF/water (1:1) gave enone 177. Protection of the hydroxyl group of 177 with tert-butyldimethylsilyl chloride and followed by bromination of the corresponding silyl ether gave bromoenone 178. Then ketalization of ketone 178 gave 179 and 180. Deprotection of ketal 179 also afforded ketal 180. Then protection of the hydroxyl group of 180 with allyl bromide afforded the allyl ether 181. Then the boronic acid 182 was prepared by a procedure similar to that for the preparation of boronic acid 167. Thus, the skeleton of 5″-epi-malyngamide C could be constructed via Suzuki cross-coupling reaction with 182 and previous prepared 172. Then the allyl ether was converted to silyl ether, followed by stereoselective epoxidation of silyl ether with hydrogen peroxide and benzyltrimethylammonium hydroxide corresponding epoxide 184. Finally, removal of the TBS protecting group with tetrabutylammonium fluoride (TBAF) provided the 5″-epi-malyngamide C, which would be convert to malyngamide C via the Mitsunobu reaction [42].

Synthesis of 5″-epi-malyngamide C reported by Cao [66]
Then malyngamide L was prepared via the similar methodology (Scheme 22) [66]. The authors initially began with (R)-(−)-carvone, which finally provided 3″,4″-epi-malyngamide K. Therefore, (S)-(−)-carvone was chosen as the starting material instead. The preparation of the enone 185 underwent a similar sequence showed in Scheme 18 in the preparation of malyngamide W. Then protection of the hydroxyl group of 185 with MOMCl gave the corresponding ether, followed by bromination to afford the bromoenone 186. Then boronic acid part 190 was prepared through the sequence as the preparation of boronic acid 182. Then Suzuki cross-coupling reaction with 190 and 172 afforded 191, which was removed allyl protecting group to finish malyngamide L.

Synthesis of malyngamide L reported by Cao [66]
4 Conclusion
This review illustrated a series of lipids which resemble anandamide in structure. At present, only several marine cyanobacterial fatty acid amides have been reported with binding affinities to the cannabinoid receptors, which were grenadamide, mooreamide, semiplenamides A, B, and G, serinolamides A, B and malyngamide B. Others, due to absence of functional assays test, only have the possibility to interact with CB1 and CB2. Additionally, the metabolites act as receptor agonists implying that they can mediate certain physiological effects through this pathway, which would open more research avenues. Further, a number of total synthesis and well-established synthetic routes have been available; these can assist structural optimization efforts towards more potent analogues, which would be of benefit for understanding the pharmacological mechanisms of cannabinoids and their receptors.
Abbreviations
- m-CPBA:
-
3-Chloroperbenzoic acid
- DCC:
-
Dicyclohexylcarbodiimide
- DDQ:
-
2, 3-Dicyano-5,6-dichlorobenzoquinone
- PMB:
-
p-Methoxybenzyl
- IBX:
-
2-Iodoxybenzoic acid
- TBDPS:
-
tert-Butyldiphenylsilyl
- TBAF:
-
Tetrabutylammonium fluoride
- TFA:
-
Trifluoroacetic acid
- MOMCl:
-
Chloromethyl methyl ether
- HMPA:
-
Hexamethylphosphoramide
- PDC:
-
Pyridinium dichlorochromate
- EDA:
-
1-Ethyl-3-(3-dimethylaminopropy)carbodiimide
References
P. Pacher, S. Bátkai, G. Kunos, Pharmacol. Rev. 58, 389–462 (2006)
A.C. Howlett, F. Barth, T.I. Bonner, G. Cabral, P. Casellas, W.A. Devane, C.C. Felder, M. Herkenham, K. Mackie, B.R. Martin, R. Mechoulam, R.G. Pertwee, Pharmacol. Rev. 54, 161–202 (2002)
C.G. Stott, G.W. Guy, Euphytica 140, 83–93 (2004)
L.A. Matsuda, S.J. Lolait, M.J. Brownstein, A.C. Young, T.I. Bonner, Nature 346, 561–564 (1990)
V.D. Marzo, L.D. Petrocellis, Annu. Rev. Med. 57, 553–574 (2006)
W.A. Devane, L. Hanus, A. Breuer, R.G. Pertwee, L.A. Stevenson, G. Griffin, D. Gibson, A. Mandelbaum, A. Etinger, R. Mechoulam, Science 258, 1946–1949 (1992)
D.M. Lambert, C.J. Fowler, J. Med. Chem. 48, 5059–5087 (2005)
V. Di Marzo, T. Bisogno, L. De Petrocellis, D. Melck, B.R. Martin, Curr. Med. Chem. 6, 721–744 (1999)
J.R. Al Dulayymi, M.S. Baird, Jones. K. Tetrahedron 60, 341–345 (2004)
E. Mevers, T. Matainaho, M. Allara, V.D. Marzo, W.H. Gerwick, Lipids 49, 1127–1132 (2014)
M. Gutiérrez, A.R. Pereira, H.M. Debonsi, A. Ligresti, V.D. Marzo, W.H. Gerwick, J. Nat. Prod. 74, 2313–2317 (2011)
R. Montaser, V.J. Paul, H. Luesch, ChemBioChem 13, 2676–2681 (2012)
B. Han, K.L. McPhail, A. Ligresti, V. Di Marzo, W.H. Gerwick, J. Nat. Prod. 66, 1364–1368 (2003)
J.W. Blunt, B.R. Copp, M.H.G. Munro, P.T. Northcote, M.R. Prinsep, Nat. Prod. Rep. 27, 165–237 (2010)
M. Nagarajan, V. Maruthanayagam, M. Sundararaman, J. Appl. Toxicol. 32, 153–185 (2012)
G.V. Reddy, T.V. Kumar, B. Siva, K.S. Babu, P.V. Srinivas, I. Sehar, A.K. Saxena, J.M. Rao, Med. Chem. Res 22, 4581–4591 (2013)
L.T. Tan, T. Okino, W.H. Gerwick, J. Nat. Prod. 63, 952–955 (2000)
M.E. Elayshberg, K.A. Blinov, A.J. Williams, E.R. Martirosian, S.G. Molodtsov, J. Nat. Prod. 65, 693–703 (2002)
S.G. Molodtsov, M.E. Elyashberg, K.A. Blinov, A.J. Williams, E.E. Martirosian, G.E. Martin, B. Lefebvre, J. Chem. Inf. Comput. Sci. 44, 1737–1751 (2004)
J.H. CardellinaII, F. Marner, R.E. Moore, J. Am. Chem. Soc. 101, 240–242 (1979)
J.H. Cardellina ll, D. Dalietos, F.-J. Marner, J.S. Mynderse, R.E. Moore, Phytochemistry 17, 2091–2095 (1978)
R.D. Ainslie, J.J. Jr Barchi, M. Kuniyoshi, R.E. Moore, J.S. Myndersel, J. Org. Chem. 50, 2859–2862 (1985)
J.S. Mynderse, R.E. Moore, J. Org. Chem. 43, 4359–4363 (1978)
W.H. Gerwick, S. Reyes, B. Alvarado, Phytochemistry 26, 1701–1704 (1987)
F.A. Villa, K. Lieske, L. Gerwick, Eur. J. Pharmacol. 629, 140–146 (2010)
A. Praud, R. Valls, L. Piovetti, B. Banaigs, Tetrahedron Lett. 34, 5437–5440 (1993)
J. Orjala, D. Nagle, W.H. Gerwick, J. Nat. Prod. 58, 764–768 (1995)
J.S. Todd, W.H. Gerwick, Tetrahedron Lett. 36, 7837–7840 (1995)
M. Wu, K.E. Milligan, W.H. Gerwick, Tetrahedron 53, 15983–15990 (1997)
Y. Kan, T. Fujita, H. Nagai, B. Sakamoto, Y. Hokama, J. Nat. Prod. 61, 152–155 (1998)
W.A. Gallimore, P.J. Scheuer, J. Nat. Prod. 63, 1422–1424 (2000)
K.E. Milligan, B. Marquez, R.T. Williamson, M. Davies-Coleman, W.H. Gerwick, J. Nat. Prod. 63, 965–968 (2000)
D.R. Appleton, M.A. Sewell, M.V. Berridge, B.R. Copp, J. Nat. Prod. 65, 630–631 (2002)
L.M. Nogle, W.H. Gerwick, J. Nat. Prod. 66, 217–220 (2003)
K.L. McPhail, W.H. Gerwick, J. Nat. Prod. 66, 132–135 (2003)
S. Suntornchashwej, K. Suwanborirux, K. Koga, M. Isobe, Chem. Asian J. 2, 114–122 (2007)
O.M. Sabry, D.E. Goeger, W.H. Gerwick, Nat. Prod. Res. 31, 555–561 (2017)
K.L. Malloy, F.A. Villa, N. Engene, T. Matainaho, W.H. Gerwick, J. Nat. Prod. 74, 95–98 (2011)
S.P. Gunasekera, C.S. Owle, R. Montaser, H. Luesch, V.J. Paul, J. Nat. Prod. 74, 871–876 (2011)
L.A. Shaala, D.T.A. Youssef, K.L. McPhail, M. Elbandy, Phytochem. Lett. 6, 183–188 (2013)
Y. Kan, B. Sakamoto, T. Fujita, H. Nagai, J. Nat. Prod. 63, 1599–1602 (2000)
J.C. Kwan, M. Teplitski, S.P. Gunasekera, V.J. Paul, H. Luesch, J. Nat. Prod. 73, 463–466 (2010)
H. Gross, K.L. Mcphall, D.E. Goeger, F.A. Valeriote, W.H. Gerwick, Phytochemistry 71, 1729–1735 (2010)
B. Han, U.M. Reinscheid, W.H. Gerwick, H. Gross, J. Mol. Struct. 989, 109–113 (2011)
F. Wan, K.L. Erickson, J. Nat. Prod. 62, 1696–1699 (1999)
R. Green, M. Cheeseman, S. Duffill, A. Merritt, S.D. Bull, Tetrahedron Lett. 46, 7931–7934 (2005)
T.D. Avery, J.A. Culbert, D.K. Taylor, Org. & Biomol. Chem. 4, 323–330 (2006)
H. Salim, O. Piva, Tetrahedron Lett. 48, 2059–2062 (2007)
T. Minuth, M.M.K. Boysen, Synthesis 16, 2799–2803 (2010)
Y.-R. Gao, S.-H. Guo, Z.-X. Zhang, S. Mao, Y.-L. Zhang, Y.-Q. Wang, Tetrahedron Lett. 54, 6511–6513 (2013)
S. Gahalawat, S.K. Pandey, RSC Adv. 5, 41013–41016 (2015)
I.R. Davies, M. Cheeseman, D.G. Niyadurupola, S.D. Bull, Tetrahedron Lett. 46, 5547–5549 (2005)
B. Das, K. Damodar, N. Bhunia, B. Shashikanth, Tetrahedron Lett. 50, 2072–2074 (2009)
M.-A. Virolleaud, C. Menant, B. Fenet, O. Piva, Tetrahedron Lett. 47, 5127–5130 (2006)
C.G. Frost, S.D. Penrosea, R. Gleave, Org. Biomol. Chem. 6, 4340–4347 (2008)
E.O.D. Oliveira, M.G. Kristin, K.P. Manoj, A. Baheti, K. Hye-Sik, L.H. MacArthur, S. Dakshanamurthy, K. Wang, L.B. Milton, M. Paige, Bioorganic & Med. Chem. 19, 4322–4329 (2011)
S. Satyanarayana, B.V.S. Reddy, R. Narender, Tetrahedron Lett. 55, 6027–6029 (2014)
J. Chen, Y. Li, X.-P. Cao, Tetrahedron Asymmetry 17, 933–941 (2006)
Y. Li, J. Chen, X.P. Cao, Synthesis 2, 320–324 (2006)
S. Suntornchashwej, K. Suwanboriruxb, M. Isobe, Tetrahedron 63, 3217–3226 (2007)
Y. Li, J.P. Feng, W.H. Wang, J. Chen, X.P. Cao, J. Org. Chem. 72, 2344–2350 (2007)
J.P. Feng, Z.F. Shi, Y. Li, J.T. Zhang, X.L. Qi, J. Chen, X.P. Cao, J. Org. Chem. 73, 6873–6876 (2008)
J. Chen, X.G. Fu, L. Zhou, J.T. Zhang, X.L. Qi, X.P. Cao, J. Org. Chem. 74, 4149–4157 (2009)
J. Chen, Z.F. Shi, L. Zhou, A.L. Xie, X.P. Cao, Tetrahedron 66, 3499–3507 (2010)
J.-T. Zhang, X.-L. Qi, J. Chen, B.-S. Li, Y.-B. Zhou, X.-P. Cao, J. Org. Chem. 76, 3946–3959 (2011)
X.L. Qi, J.T. Zhang, J.P. Feng, X.P. Cao, Org. Biomol. Chem. 9, 3817–3824 (2011)
Acknowledgments
We are grateful for financial support from National Natural Science Foundation of China (NSFC-21572178 and NSFC-21702162).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Gao, YR., Wang, YQ. Cannabinomimetric Lipids: From Natural Extract to Artificial Synthesis. Nat. Prod. Bioprospect. 8, 1–21 (2018). https://doi.org/10.1007/s13659-017-0151-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13659-017-0151-9