
Abstract
Atopic dermatitis (AD) is a chronic, relapsing immunoinflammatory skin condition characterized by sensations such as pruritis, pain, and neuronal hypersensitivity. The mechanisms underlying these sensations are multifactorial and involve complex crosstalk among several cutaneous components. This review explores the role these components play in the pathophysiology of atopic dermatitis. In the skin intercellular spaces, sensory nerves interact with keratinocytes and immune cells via myriad mediators and receptors. These interactions generate action potentials that transmit pruritis and pain signals from the peripheral nervous system to the brain. Keratinocytes, the most abundant cell type in the epidermis, are key effector cells, triggering crosstalk with immune cells and sensory neurons to elicit pruritis, pain, and inflammation. Filaggrin expression by keratinocytes is reduced in atopic dermatitis, causing a weakened skin barrier and elevated skin pH. Fibroblasts are the main cell type in the dermis and, in atopic dermatitis, appear to reduce keratinocyte differentiation, further weakening the skin barrier. Fibroblasts and mast cells promote inflammation while dermal dendritic cells appear to attenuate inflammation. Inflammatory cytokines and chemokines play a major role in AD pathogenesis. Type 2 immune responses typically generate pruritis, and the type 1 and type 3 responses generate pain. Type 2 responses and increased skin pH contribute to barrier dysfunction and promote dysbiosis of the skin microbiome, causing the proliferation of Staphyloccocus aureus. In conclusion, understanding the dynamic interactions between cutaneous components in AD could drive the development of therapies to improve the quality of life for patients with AD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Atopic dermatitis (AD) is a chronic skin disease characterized by sensations such as pruritis and pain due to neuronal hypersensitivity. |
The mechanisms underlying AD sensations are multifactorial and involve complex crosstalk among several cutaneous components including peripheral sensory neurons, keratinocytes, infiltrating immune cells, and the skin microbiome. |
As the most abundant cell type in the epidermis, keratinocytes are key effector cells, triggering crosstalk with immune cells and sensory neurons to elicit itching, pain, and inflammation. |
Understanding the dynamic molecular, cellular, and microbial interactions in AD could drive the development of new therapeutic approaches to relieving cutaneous pain and pruritis. |
Introduction
Atopic dermatitis (AD) is a chronic, relapsing immunoinflammatory skin condition characterized by localized eczema, scaly dry skin, and intense pruritis [1, 2]. Disease onset typically manifests during childhood, but incident disease may also occur during adulthood [3]. AD affects approximately 0.96–22.6% of children and 1.2–17.1% of adults, and the incidence appears to be increasing in industrialized countries [2, 4]. AD is associated with a profound negative impact on the psychosocial functioning and quality of life of patients, affecting sleep, social interactions, mental health, and work productivity [5, 6]. Patients with AD often identify pruritis and pain/soreness as the most important symptoms when assessing treatment efficacy [7].
AD has heterogeneous clinical characteristics, but intense itch, defined as an unpleasant sensory perception that causes a desire to scratch, is the predominant feature [8,9,10]. Chronic itch, associated with inflammation in AD, is defined as a continuous pruritis lasting longer than 6 weeks [11]. The mechanisms leading to itching in AD and an itch–scratch cycle are multifactorial and involve crosstalk between the skin microbiome, the epidermal barrier, keratinocytes, immune cells, and sensory nerves [3].
Skin pain is frequently described in AD and affects between 42.7% and 92.2% of patients with AD [10, 12,13,14,15]. Some patients experience only pain or itch, but 59–78% of patients with AD experience these sensations concomitantly [14, 16, 17]. Patients often describe their pruritis as “tingling,” “burning,” “searing,” and “stinging,” terms that also describe neuropathic pain [12, 15, 17, 18]. The mechanisms of pain in AD are still poorly understood compared with pruritis but likely involve nociceptive pain caused by the activation of peripheral nerve fibers by scratching and inflammatory pain associated with tissue damage and inflammation [19,20,21].
In patients with AD who experience chronic inflammation and tissue damage, a neural sensitization phenomenon is also noted [17, 22, 23]. Chronic exposure to inflammatory mediators or trauma (such as scratching) associated with AD can cause primary sensory neuron afferents and central synapses to amplify information and become hyper-responsive over time [23]. Central and peripheral neuronal sensitization can lead to hyperknesis (increased sensitivity of nerves to pruritic stimuli) and alloknesis (nonpruritic stimuli are perceived as itch), which further contribute to the chronic nature of AD [17, 23,24,25].
The pathophysiology of pruritis, pain, and neurosensitization in AD is complex and involves crosstalk between several components, including the peripheral and central nervous system, resident epidermal skin cells, immune cells, and the skin microbiome [3, 23, 25]. Here, we review the role of cutaneous components in the mechanisms mediating pruritis, pain, and neurosensitivity in AD.
Methods
A literature review was performed using PubMed to identify relevant primary research and review articles published between 2010 and 2023. Seminal articles published prior to 2010 were included as appropriate. This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Overview of Cutaneous Components
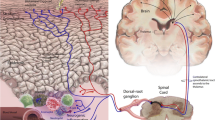
The epidermis, dermis, and hypodermis are the main three layers of the skin (Fig. 1) [26]. The superficial component of the skin is the epidermis, which consists of several layers of keratinocytes in progressive stages of differentiation following their germination at the stratum basal layer. The stratum corneum (SC) is the outermost layer of the epidermis and forms the skin’s first physical barrier against water loss and penetration by environmental irritants. The SC also provides an environment for commensal skin microbiome colonization. Below the SC are several layers of keratinocytes interspersed with Langerhans cells (LCs), melanocytes, and Merkel cells (MCs), as well as infiltrating immune cells. Below the epidermis, separated by a basement membrane, is the dermis containing infiltrating immune cells, fibroblasts, mast cells, dermal dendritic cells, peripheral sensory neurons, blood and lymphatic vessels, hair follicles, and sweat glands. The hypodermis is the innermost layer of skin and contains large amounts of adipocytes and fibroblasts [26, 27]. Figure 1 illustrates a schematic of the skin indicating the different cutaneous components contributing to pruritis, pain, and neurosensitization in AD.

The skin is a laminated structure composed of several components that contribute to pruritis, pain, and neurosensitization in atopic dermatitis: the neurosensory component (nociceptors and pruriceptors), resident cell component (keratinocytes, Langerhans cells, Merkel cells, fibroblasts, mast cells), immune/infiltrating cell component (macrophages and lymphocytes), and the skin microbiome
Neurosensory Component
Sensory nerves innervate all skin layers and create a complex neural network that extends to the stratum corneum [11, 25, 28]. In the skin intercellular spaces, sensory nerves come in close contact with resident (e.g., keratinocytes and dendritic cells) and infiltrating cells (e.g., lymphocytes and eosinophils) and interact with these via myriad mediators and receptors [11, 20, 22, 25]. These interactions generate action potentials that transmit signals from the peripheral nervous system to the dorsal root and trigeminal ganglia, where they are then conveyed to the spinal cord and, ultimately, the brain for interpretation [23, 25].
Pruriceptive neurons are a subset of nociceptors that can be activated by itch-generating mechanical or chemical stimuli [29]. Most pruriceptors are polymodal and can be activated by capsaicin, a classical pain stimulus [20, 29,30,31]. It is thought that noxious stimuli selectively activate distinct receptor and intracellular signaling pathways in the same neurons for transducing pruritis or pain signaling [20, 30].
Pruritis in AD is primarily perceived via nonhistaminergic sensory nerves that can be activated via receptors for endogenous and exogenous pruritogens other than histamine, such as proteases, cytokines, and amines [11, 23, 32]. Nonhistamine receptors of importance in AD include cytokine receptors and various G protein-coupled receptors other than histamine receptors, such as protease-activated receptors (PARs), toll-like receptors (TLRs), and Mas-related G protein-coupled receptors (Mrgprs) [20, 30, 32]. Genetic and functional analyses initially supported the existence of itch-specific neurons that express Mrgprs [33]. MrgprX2 is upregulated in patients with AD and correlates with itch intensity [34].
The generation and transmission of neurosensory signals depend on ion channel activation to generate action potentials. Both itch- and pain-sensory neurons employ many of the same ion channels to transmit their signals, including the voltage-gated sodium channels NaV1.7 and NaV1.8, transient receptor potential ankyrin 1 (TRPA1), and transient receptor potential vanilloid 1 (TRPV1) [8, 35]. Recent evidence suggests that the function of PIEZO ion channels may extend beyond mechanotransduction; PIEZO1 plays a role in chronic pruritis but not pain in mice, and PIEZO2 may contribute to both alloknesis and pain pathogenesis [36,37,38].
An imbalance between nerve elongation and repulsion factors (such as nerve growth factor and semaphorin 3A, respectively) in the skin of patients with AD has the potential to enhance nerve sprouting and promote enhanced branching of intraepidermal nerves [39, 40]. This might contribute to neural sensitization by lowering the activation threshold of sensory neurons [25, 40]. However, there is conflicting evidence regarding the density of nerve fibers in human AD skin; it is not clear whether hyperinnervation plays a role in AD pathogenesis [41,42,43,44,45].
Resident and Infiltrating Cell Components
Keratinocytes
Keratinocytes comprise most cells in the epidermis and are involved in skin barrier functions [46]. The function of the epidermis as a physical barrier depends primarily on the outermost epidermal layer, the SC. It is the end product of the progressive differentiation of keratinocytes and comprises 10–20 layers of corneal keratinocytes embedded in a complex hydrophobic extracellular matrix consisting of ceramides, fatty acids, cholesterol lipids, and proteins [27, 47]. Keratinocytes express several structural proteins as they mature, including loricrin, involucrin, and filaggrin (FLG), which is the main structural protein in the SC [48]. Genetic studies have demonstrated a strong association between AD and several loss-of-function mutations in the filaggrin gene (FLG). Filaggrin deficiency in AD also alters the acidic milieu of the SC, with reduced urocanic acid being a possible cause of elevated skin pH in AD [49].
Keratinocytes are considered the key effector cells in AD, triggering crosstalk with immune cells and sensory neurons to elicit pruritis and inflammation [46, 50]. They also express various receptors involved in pruritis and pain also found on nerve fibers, including protease-activated receptor 2 (PAR2), aryl hydrocarbon receptors (AhRs), transient receptor potential ion channels (including TRPV1, TRPV3, and TRPA1), tropomyosin receptor kinases A and B (TrkA and TrkB), interleukin (IL)-31 receptors, and μ and κ opioid receptors [28, 51, 52]. Therefore, keratinocytes themselves can be activated by binding pruriceptive and algogenic molecules to surface receptors. For example, keratinocytes release thymic stromal lymphopoietin (TSLP) in response to a range of stimuli, including protease activation of PAR2. TSLP activates other immune cells but can also directly stimulate pruriceptive sensory nerve fibers to induce pruritis, a finding that has also been shown for the alarmin IL-33 [25, 35, 53]. Of the protease alarmins released by keratinocytes, various kallikreins (KLKs), including KLK5, KLK7, KLK8, and KLK13, are elevated in lesional AD skin [34, 54]. KLK7 contributes to chronic pruritis independent of skin inflammation [54].
Periostin is a multifunctional extracellular matrix protein that is highly expressed in both keratinocytes and fibroblasts in the dermis and plays a role in skin inflammation and pruritis via the activation of integrin α-IIb on sensory nerves and amplification of the secretion of type 2 cytokines [55, 56]. The release of periostin from keratinocytes and fibroblasts is further stimulated by TSLP and other type 2 cytokines, such as IL-4 and IL-13, thus inducing a positive feedback cycle of inflammation [40, 56].
Keratinocytes release many other molecules, including nerve growth factor (NGF), opioids, substance P, neurotrophin 4, endocannabinoids, artemin, and acetylcholine, which have been implicated in pruritis and/or pain in AD [28, 57]. For example, NGF binds to TrkA on nociceptors to initiate both pruritis and pain signaling [32, 58, 59]. Acetylcholine can activate sensory nerves both directly and indirectly, the latter by lowering the activation threshold for other stimuli [28, 57].
Merkel Cells
MCs are situated within the basal layer of the epidermis and are closely associated with low-threshold, slowly adapting Aβ sensory fibers, which together form Merkel discs. MCs are primarily mechanosensory cells and may play a role in alloknesis, but generally, the role these cells play in chronic pruritis and/or pain in AD remains unclear [50]. The mechanosensitive PIEZO1 ion channel is expressed in MCs, and recent evidence suggests that PIEZO1 may play a role in chronic pruritis but not pain, while PIEZO2 may contribute to alloknesis and pain [36,37,38].
Fibroblasts
Fibroblasts are the principal cell type in the dermis and exhibit considerable heterogeneity [60] Using single-cell transcriptome analysis, He et al. found a novel fibroblast subpopulation that was unique to lesional AD skin and expressed C–C chemokine ligand 2 (CCL2) and CCL19 [61]. These researchers also found a corresponding dendritic cell population that expressed C–C chemokine receptor type 7, the CCL19 receptor, thereby demonstrating possible crosstalk between fibroblasts and immune cells in AD. In AD skin, fibroblasts have been shown to impair the proliferation of keratinocytes and the terminal differentiation process, partially due to reduced expression of the differentiation-associated cytokine leukemia inhibitory factor by atopic fibroblasts [62]. The exact role of fibroblasts in pruritis and pain pathogenesis in AD remains to be determined.
Mast Cells
Mast cells are tissue-resident immune cells filled with cytosolic granules. Upon activation, mast cells undergo degranulation and release the content of their cytosolic granules containing histamine, serotonin, leukotriene, proteases, cytokines, and chemokines [30, 63,64,65] Mast cell activation also prompts the de novo synthesis of many cytokines and chemokines as well as lipid mediators such as prostanoids [65, 66]. Proteases produced by mast cells, such as tryptases, chymases, cathepsins, and KLKs, can induce strong nonhistaminergic pruritis by binding to PARs on keratinocytes and sensory neurons [32, 67]. Activation of MrgprX2 on mast cells has been found to cause nonhistaminergic pruritis [68]. MrgprX1 and MrgprX2 have also been implicated in neuropathic and inflammatory pain via mast cell activation [69, 70]. Targeting mast cell-associated Mrgprs may offer promising therapies for AD.
Dendritic Cells
Dendritic cells represent a heterogeneous family of bone marrow-derived leukocytes that link innate and adaptive immunity [71]. LCs are a unique population of tissue-resident dendritic cells that form a network of cells across the epidermis [72]. Upon activation, LCs migrate to regional lymph nodes, where they prime T lymphocytes to induce an immune response [73, 74].
Although more LCs are present and reside in an activated state in AD lesions, the exact role they play in this disease is unclear [73]. Some evidence demonstrates that TSLP causes the proliferation of skin-resident LCs but not monocyte-derived LCs. A defective epidermal barrier in AD possibly causes altered LC behavior, including increased proliferation rates and an enhanced activation state that instigates inflammation in AD. The high-affinity immunoglobulin E (IgE) receptor is expressed at higher levels on LCs in AD lesions, and the binding of IgE molecules to the surface of LCs enhances antigen uptake [73, 74].
Although LCs are the main dendritic cell in healthy skin, AD skin is additionally colonized by inflammatory dendritic epidermal cells (IDECs) that contribute to ongoing inflammation pathology [75]. Both LCs and IDECs express several TLRs to sense danger signals from the external environment, such as from the skin microbiome, and TLR prompts the activation and maturation of dendritic cells, characterized by the expression of costimulatory molecules and secretion of cytokines. In particular, TLR2 stimulation due to a lipopeptide from the commensal bacterium Staphylococcus epidermidis is known to prompt CD36–p38 mitogen-activated protein kinase signaling in keratinocytes to increase antibacterial defense; however, TLR2 expression is decreased in epidermal LC and IDEC [76, 77]. This may reduce the inflammatory response, thus contributing to heightened S. aureus colonization in patients with AD [78, 79].
Immune Component
Inflammatory cytokines and chemokines play a major role in AD pathogenesis, and pruritis and pain pathways are immunologically distinct [35]. Cytokines associated with type 1 and/or type 3 immune responses, such as IL-1 β, IL-6, IL-17A and tumor necrosis factor α, together with chemokines, such as CCL2, CCL5, and C–X–C motif chemokine 1, have been associated with pain [8, 35, 80, 81]. Pruritis in AD is associated with increased type 2 cytokines, such as IL-4, IL-13, and IL-31 [35, 81]. Macrophages have also been implicated as cellular sources of IL-31 [82].
Activated keratinocytes significantly increase the release of proinflammatory cytokines such as IL-1, IL-25, IL-33, and TSLP. These cytokines activate LCs and dendritic cells in the epidermis and dermis, leading to the activation of T helper 2 cells, which, in turn, causes the production of more inflammatory cytokines and further activation of keratinocytes [46]. IL-31 is a key cytokine mediating pruritis in AD by binding to its receptor complex, comprising an IL-31 receptor A and oncostatin M receptor, expressed on peripheral nerve fibers, dorsal root ganglia, and keratinocytes [40, 83]. IL-31 can also generate morphological changes in neurons that increase sensitivity [84]. IL-4 and IL-13 induce chronic pruritis in a mouse model of AD by activating sensory neurons through the IL-4 receptor α and Janus kinase 1 [85].
Type 2 immune responses contribute to barrier dysfunction in AD. For example, the expression of genes regulating keratin, filaggrin, involucrin, and loricrin production is downregulated by IL-4, IL-13, and IL-22, thereby suppressing the development of the filaggrin/keratin structural network [86,87,88]. IL-4 and IL-13 also significantly induce keratinocytes to release the peptidase KLK7, which induces pruritis in AD without inflammation and degrades corneodesmosomal proteins to initiate skin desquamation [54, 88]. Type 2 responses also negatively influence ceramide synthesis in the SC [86].
IL-4 contributes to S. aureus colonization of the disrupted skin barrier by suppressing the production of antimicrobial peptides (AMP) by keratinocytes and triggering fibroblast production of collagen, fibronectin, and fibrinogen, which serve as adhesion molecules for S. aureus [87].
Microbial Component
In healthy skin, the microbiome plays an important role in inhibiting pathogen colonization and growth, such as by the secretion of AMPs, and modulates innate and adaptive immune responses [89]. For example, skin microbiota produce ligands, including tryptophan metabolites and short-chain fatty acids, that activate the aryl hydrocarbon receptor to initiate host defense responses by keratinocytes [52].
In AD, increased skin pH promotes microbial dysbiosis, with a reduction of microbial diversity and proliferation of S. aureus [78, 79, 90, 91]. Increased pH in atopic skin also increases the expression of secreted and cell wall-associated proteins involved in immune evasion and adherence, such as clumping factor B and fibronectin-binding protein, which further promotes S. aureus colonization on skin affected by AD [79].
Elevated skin pH and the proliferation of S. aureus in AD promote an increased abundance of microbiome-derived serine proteases in lesional skin [92]. These proteases induce pruritis by activating PAR2 and PAR4 receptors in keratinocytes and pruriceptors [32, 78]. Staphylococcus δ-toxin activates MrgprX2 to induce mast cell degranulation, which induces the release of several proinflammatory mediators including histamine, ILs, tumor necrosis factor, and prostaglandin D2 [93]. MrgprX2 is also activated by AMPs, produced in response to stimuli such as microbial pathogens, to promote the release of IL-31 [78].
Studies of pain during S. aureus infection have indicated that pathogens can directly activate nociceptors through the release of N-formyl peptides and the pore-forming toxin α hemolysin and modulation of ion channel activity [94].
Conclusions
Both pruritis and pain are common symptoms in patients with AD, and patients may show neuronal sensitization to pruritis and pain. Multiple cutaneous components play a role in the pathophysiology of AD and understanding the specific pathways involved in pruritis, pain, and neurosensitivity may offer new insights regarding the correlation between available therapies and pruritis/pain relief. Both pruritis and pain are common in patients with AD, and associated neuronal sensitization foments these symptoms. Beyond the components of the sensory nervous system, multiple additional cutaneous elements play a role in the pathophysiology of AD.
Several new therapeutics show promise for treating the symptoms of itch and pain in patients with AD. Topical phosphodiesterase 4 inhibitors have been shown to reduce levels of IL-4 and IL-31, which are key cytokines governing pruritis in AD [81, 95]. Clinically meaningful improvements in pruritis have been observed with Janus kinase (JAK) inhibitors, including the JAK1 selective inhibitor, abrocitinib [96]. JAK inhibitors alleviate pruritis by suppressing the JAK signaling pathway, which is involved in the production of proinflammatory cytokines [97, 98]. However, some JAK inhibitors, such as tofacitinib and ruxolitinib, are known to cause application site discomfort (pain and burning sensations) [97]. The monoclonal antibody dupilumab targets the α subunit of the IL-4 and IL-13 receptor complex, which has a known role in pruritis in AD [81, 99]. Dupilumab has been shown to reduce pruritis in AD, but its role in pain is unknown [99]. As our understanding of the dynamic molecular, cellular, and microbial interactions in normal and skin affected by AD increases, it is reasonable to expect that these developments will lead to new insights into the similarities and differences between chronic pruritis and chronic pain in AD.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
References
Boguniewicz M, Fonacier L, Guttman-Yassky E, Ong PY, Silverberg J, Farrar JR. Atopic dermatitis yardstick: practical recommendations for an evolving therapeutic landscape. Ann Allergy Asthma Immunol. 2018;120:10-22.e2.
Ständer S. Atopic dermatitis. N Engl J Med. 2021;384:1136–43.
Langan SM, Irvine AD, Weidinger S. Atopic dermatitis. Lancet. 2020;396:345–60.
Bylund S, Kobyletzki LB, Svalstedt M, Svensson Å. Prevalence and incidence of atopic dermatitis: a systematic review. Acta Derm Venereol. 2020;100:adv00160.
Murota H, Koike Y, Morisaki H, Matsumoto M, Takenaka M. Exacerbating factors and disease burden in patients with atopic dermatitis. Allergol Int. 2022;71:25–30.
Na SY, Moon W. Perspectives on current and novel treatments for inflammatory bowel disease. Gut Liver. 2019;13:604–16.
von Kobyletzki LB, Thomas KS, Schmitt J, Chalmers JR, Deckert S, Aoki V, et al. What factors are important to patients when assessing treatment response: an international cross-sectional survey. Acta Derm Venereol. 2017;97:86–90.
Li JX, Dong RJ, Zeng YP. Characteristics, mechanism, and management of pain in atopic dermatitis: a literature review. Clin Transl Allergy. 2021;11: e12079.
Weidinger S, Beck LA, Bieber T, Kabashima K, Irvine AD. Atopic dermatitis. Nat Rev Dis Primers. 2018;4:1.
Misery L, Belloni Fortina A, El Hachem M, Chernyshov P, von Kobyletzki L, Heratizadeh A, et al. A position paper on the management of itch and pain in atopic dermatitis from the international society of atopic dermatitis (isad)/oriented patient-education network in dermatology (opened) task force. J Eur Acad Dermatol Venereol. 2021;35:787–96.
Azimi E, Xia J, Lerner EA. Peripheral mechanisms of itch. Curr Probl Dermatol. 2016;50:18–23.
Vakharia PP, Chopra R, Sacotte R, Patel KR, Singam V, Patel N, et al. Burden of skin pain in atopic dermatitis. Ann Allergy Asthma Immunol. 2017;119(548–52): e3.
Huet F, Shourick J, Séité S, Taïeb C, Misery L. Pain in atopic dermatitis: an online population-based survey. Acta Derm Venereol. 2020;100:adv00198.
Dawn A, Papoiu AD, Chan YH, Rapp SR, Rassette N, Yosipovitch G. Itch characteristics in atopic dermatitis: results of a web-based questionnaire. Br J Dermatol. 2009;160:642–4.
Pojawa-Gołąb M, Reich A. Skin pain in patients with atopic dermatitis or psoriasis: a web-based survey. Acta Derm Venereol. 2020;100:adv00258.
Maarouf M, Kromenacker B, Capozza KL, Kempton D, Hendricks A, Tran K, et al. Pain and itch are dual burdens in atopic dermatitis. Dermatitis. 2018;29:278–81.
Andersen HH, Elberling J, Sølvsten H, Yosipovitch G, Arendt-Nielsen L. Nonhistaminergic and mechanical itch sensitization in atopic dermatitis. Pain. 2017;158:1780–91.
Rosen JD, Fostini AC, Chan YH, Nattkemper LA, Yosipovitch G. Cross-sectional study of clinical distinctions between neuropathic and inflammatory pruritus. J Am Acad Dermatol. 2018;79:1143–4.
Woolf CJ. What is this thing called pain? J Clin Invest. 2010;120:3742–4.
Liu T, Ji RR. New insights into the mechanisms of itch: are pain and itch controlled by distinct mechanisms? Pflugers Arch. 2013;465:1671–85.
Silverberg JI, Gelfand JM, Margolis DJ, Boguniewicz M, Fonacier L, Grayson MH, et al. Pain is a common and burdensome symptom of atopic dermatitis in united states adults. J Allergy Clin Immunol Pract. 2019;7(2699–706): e7.
Tominaga M, Takamori K. Peripheral itch sensitization in atopic dermatitis. Allergol Int. 2022;71:265–77.
Yosipovitch G, Berger T, Fassett MS. Neuroimmune interactions in chronic itch of atopic dermatitis. J Eur Acad Dermatol Venereol. 2020;34:239–50.
Ikoma A, Steinhoff M, Ständer S, Yosipovitch G, Schmelz M. The neurobiology of itch. Nat Rev Neurosci. 2006;7:535–47.
Legat FJ. Itch in atopic dermatitis—what is new? Front Med (Lausanne). 2021;8: 644760.
Salimian J, Salehi Z, Ahmadi A, Emamvirdizadeh A, Davoudi SM, Karimi M, et al. Atopic dermatitis: Molecular, cellular, and clinical aspects. Mol Biol Rep. 2022;49:3333–48.
Lawton S. Skin 1: The structure and functions of the skin. Nurs Times. 2019;115:30–3.
Mollanazar NK, Smith PK, Yosipovitch G. Mediators of chronic pruritus in atopic dermatitis: getting the itch out? Clin Rev Allergy Immunol. 2016;51:263–92.
LaMotte RH, Dong X, Ringkamp M. Sensory neurons and circuits mediating itch. Nat Rev Neurosci. 2014;15:19–31.
Dong X, Dong X. Peripheral and central mechanisms of itch. Neuron. 2018;98:482–94.
Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–24.
Yosipovitch G, Rosen JD, Hashimoto T. Itch: from mechanism to (novel) therapeutic approaches. J Allergy Clin Immunol. 2018;142:1375–90.
Han L, Ma C, Liu Q, Weng HJ, Cui Y, Tang Z, et al. A subpopulation of nociceptors specifically linked to itch. Nat Neurosci. 2013;16:174–82.
Nattkemper LA, Tey HL, Valdes-Rodriguez R, Lee H, Mollanazar NK, Albornoz C, et al. The genetics of chronic itch: gene expression in the skin of patients with atopic dermatitis and psoriasis with severe itch. J Invest Dermatol. 2018;138:1311–7.
Trier AM, Mack MR, Kim BS. The neuroimmune axis in skin sensation, inflammation, and immunity. J Immunol. 2019;202:2829–35.
Hill RZ, Loud MC, Dubin AE, Peet B, Patapoutian A. Piezo1 transduces mechanical itch in mice. Nature. 2022;607:104–10.
Shin SM, Moehring F, Itson-Zoske B, Fan F, Stucky CL, Hogan QH, et al. Piezo2 mechanosensitive ion channel is located to sensory neurons and nonneuronal cells in rat peripheral sensory pathway: implications in pain. Pain. 2021;162:2750–68.
Feng J, Luo J, Yang P, Du J, Kim BS, Hu H. Piezo2 channel-merkel cell signaling modulates the conversion of touch to itch. Science. 2018;360:530–3.
Tominaga M, Ogawa H, Takamori K. Decreased production of semaphorin 3a in the lesional skin of atopic dermatitis. Br J Dermatol. 2008;158:842–4.
Nakahara T, Kido-Nakahara M, Tsuji G, Furue M. Basics and recent advances in the pathophysiology of atopic dermatitis. J Dermatol. 2021;48:130–9.
Guseva D, Rüdrich U, Kotnik N, Gehring M, Patsinakidis N, Agelopoulos K, et al. Neuronal branching of sensory neurons is associated with bdnf-positive eosinophils in atopic dermatitis. Clin Exp Allergy. 2020;50:577–84.
Sugiura H, Omoto M, Hirota Y, Danno K, Uehara M. Density and fine structure of peripheral nerves in various skin lesions of atopic dermatitis. Arch Dermatol Res. 1997;289:125–31.
Urashima R, Mihara M. Cutaneous nerves in atopic dermatitis. A histological, immunohistochemical and electron microscopic study. Virchows Arch. 1998;432:363–70.
Tsutsumi M, Kitahata H, Fukuda M, Kumamoto J, Goto M, Denda S, et al. Numerical and comparative three-dimensional structural analysis of peripheral nerve fibres in epidermis of patients with atopic dermatitis. Br J Dermatol. 2016;174:191–4.
Tan Y, Ng WJ, Lee SZX, Lee BTK, Nattkemper LA, Yosipovitch G, et al. 3-dimensional optical clearing and imaging of pruritic atopic dermatitis and psoriasis skin reveals downregulation of epidermal innervation. J Invest Dermatol. 2019;139:1201–4.
Das P, Mounika P, Yellurkar ML, Prasanna VS, Sarkar S, Velayutham R, et al. Keratinocytes: An enigmatic factor in atopic dermatitis. Cells. 2022;11:1683.
Haftek M. Epidermal barrier disorders and corneodesmosome defects. Cell Tissue Res. 2015;360:483–90.
Furue M. Regulation of filaggrin, loricrin, and involucrin by il-4, il-13, il-17a, il-22, ahr, and nrf2: pathogenic implications in atopic dermatitis. Int J Mol Sci. 2020;21:5382.
Moosbrugger-Martinz V, Leprince C, Méchin MC, Simon M, Blunder S, Gruber R, et al. Revisiting the roles of filaggrin in atopic dermatitis. Int J Mol Sci. 2022;23:5318.
Agelopoulos K, Pereira MP, Wiegmann H, Ständer S. Cutaneous neuroimmune crosstalk in pruritus. Trends Mol Med. 2022;28:452–62.
Larkin C, Chen W, Szabó IL, Shan C, Dajnoki Z, Szegedi A, et al. Novel insights into the trpv3-mediated itch in atopic dermatitis. J Allergy Clin Immunol. 2021;147:1110-4.e5.
van den Bogaard EH, Esser C, Perdew GH. The aryl hydrocarbon receptor at the forefront of host-microbe interactions in the skin: a perspective on current knowledge gaps and directions for future research and therapeutic applications. Exp Dermatol. 2021;30:1477–83.
Wilson SR, Thé L, Batia LM, Beattie K, Katibah GE, McClain SP, et al. The epithelial cell-derived atopic dermatitis cytokine tslp activates neurons to induce itch. Cell. 2013;155:285–95.
Guo CJ, Mack MR, Oetjen LK, Trier AM, Council ML, Pavel AB, et al. Kallikrein 7 promotes atopic dermatitis-associated itch independently of skin inflammation. J Invest Dermatol. 2020;140:1244-52.e4.
Hashimoto T, Mishra SK, Olivry T, Yosipovitch G. Periostin, an emerging player in itch sensation. J Invest Dermatol. 2021;141:2338–43.
Mishra SK, Wheeler JJ, Pitake S, Ding H, Jiang C, Fukuyama T, et al. Periostin activation of integrin receptors on sensory neurons induces allergic itch. Cell Rep. 2020;31: 107472.
Steinhoff M, Ahmad F, Pandey A, Datsi A, AlHammadi A, Al-Khawaga S, et al. Neuroimmune communication regulating pruritus in atopic dermatitis. J Allergy Clin Immunol. 2022;149:1875–98.
Yu S, Zhou Y, Follansbee T, Hwang ST. Immune mediators and therapies for pruritus in atopic dermatitis and psoriasis. J Cutane Immunol All. 2019;2:4–14.
Hefti FF, Rosenthal A, Walicke PA, Wyatt S, Vergara G, Shelton DL, et al. Novel class of pain drugs based on antagonism of ngf. Trends Pharmacol Sci. 2006;27:85–91.
Muhl L, Genové G, Leptidis S, Liu J, He L, Mocci G, et al. Single-cell analysis uncovers fibroblast heterogeneity and criteria for fibroblast and mural cell identification and discrimination. Nat Commun. 2020;11:3953.
He H, Suryawanshi H, Morozov P, Gay-Mimbrera J, Del Duca E, Kim HJ, et al. Single-cell transcriptome analysis of human skin identifies novel fibroblast subpopulation and enrichment of immune subsets in atopic dermatitis. J Allergy Clin Immunol. 2020;145:1615–28.
Berroth A, Kühnl J, Kurschat N, Schwarz A, Stäb F, Schwarz T, et al. Role of fibroblasts in the pathogenesis of atopic dermatitis. J Allergy Clin Immunol. 2013;131:1547–54.
Levi-Schaffer F, Gibbs BF, Hallgren J, Pucillo C, Redegeld F, Siebenhaar F, et al. Selected recent advances in understanding the role of human mast cells in health and disease. J Allergy Clin Immunol. 2022;149:1833–44.
Schuler CF, Billi AC, Maverakis E, Tsoi LC, Gudjonsson JE. Novel insights into atopic dermatitis. J Allergy Clin Immunol. 2022;151:1145–54.
Wernersson S, Pejler G. Mast cell secretory granules: armed for battle. Nat Rev Immunol. 2014;14:478–94.
Honda T, Kabashima K. Prostanoids and leukotrienes in the pathophysiology of atopic dermatitis and psoriasis. Int Immunol. 2019;31:589–95.
Akiyama T, Lerner EA, Carstens E. Protease-activated receptors and itch. Handb Exp Pharmacol. 2015;226:219–35.
Meixiong J, Anderson M, Limjunyawong N, Sabbagh MF, Hu E, Mack MR, et al. Activation of mast-cell-expressed mas-related g-protein-coupled receptors drives non-histaminergic itch. Immunity. 2019;50:1163-71.e5.
Green DP. The role of mrgprs in pain. Neurosci Lett. 2021;744:135544.
Navratilova E, Porreca F. Substance p and inflammatory pain: getting it wrong and right simultaneously. Neuron. 2019;101:353–5.
Shortman K, Liu YJ. Mouse and human dendritic cell subtypes. Nat Rev Immunol. 2002;2:151–61.
West HC, Bennett CL. Redefining the role of langerhans cells as immune regulators within the skin. Front Immunol. 2017;8:1941.
Deckers J, Hammad H, Hoste E. Langerhans cells: sensing the environment in health and disease. Front Immunol. 2018;9:93.
Rajesh A, Wise L, Hibma M. The role of langerhans cells in pathologies of the skin. Immunol Cell Biol. 2019;97:700–13.
Wollenberg A, Kraft S, Hanau D, Bieber T. Immunomorphological and ultrastructural characterization of langerhans cells and a novel, inflammatory dendritic epidermal cell (idec) population in lesional skin of atopic eczema. J Invest Dermatol. 1996;106:446–53.
Iwamoto K, Nümm TJ, Koch S, Herrmann N, Leib N, Bieber T. Langerhans and inflammatory dendritic epidermal cells in atopic dermatitis are tolerized toward tlr2 activation. Allergy. 2018;73:2205–13.
Li D, Lei H, Li Z, Li H, Wang Y, Lai Y. A novel lipopeptide from skin commensal activates tlr2/cd36-p38 mapk signaling to increase antibacterial defense against bacterial infection. PLoS ONE. 2013;8: e58288.
Li W, Yosipovitch G. The role of the microbiome and microbiome-derived metabolites in atopic dermatitis and non-histaminergic itch. Am J Clin Dermatol. 2020;21:44–50.
Koh LF, Ong RY, Common JE. Skin microbiome of atopic dermatitis. Allergol Int. 2022;71:31–9.
Kiguchi N, Kobayashi Y, Kishioka S. Chemokines and cytokines in neuroinflammation leading to neuropathic pain. Curr Opin Pharmacol. 2012;12:55–61.
Kwatra SG, Misery L, Clibborn C, Steinhoff M. Molecular and cellular mechanisms of itch and pain in atopic dermatitis and implications for novel therapeutics. Clin Transl Immunology. 2022;11: e1390.
Hashimoto T, Yokozeki H, Karasuyama H, Satoh T. Il-31-generating network in atopic dermatitis comprising macrophages, basophils, thymic stromal lymphopoietin, and periostin. J Allergy Clin Immunol. 2023;151:737-46.e6.
Furue M, Yamamura K, Kido-Nakahara M, Nakahara T, Fukui Y. Emerging role of interleukin-31 and interleukin-31 receptor in pruritus in atopic dermatitis. Allergy. 2018;73:29–36.
Feld M, Garcia R, Buddenkotte J, Katayama S, Lewis K, Muirhead G, et al. The pruritus- and th2-associated cytokine il-31 promotes growth of sensory nerves. J Allergy Clin Immunol. 2016;138(500–8): e24.
Oetjen LK, Mack MR, Feng J, Whelan TM, Niu H, Guo CJ, et al. Sensory neurons co-opt classical immune signaling pathways to mediate chronic itch. Cell. 2017;171(217–28):e13.
Beck LA, Cork MJ, Amagai M, De Benedetto A, Kabashima K, Hamilton JD, et al. Type 2 inflammation contributes to skin barrier dysfunction in atopic dermatitis. JID Innov. 2022;2:100131.
Chiricozzi A, Maurelli M, Peris K, Girolomoni G. Targeting il-4 for the treatment of atopic dermatitis. Immunotargets Ther. 2020;9:151–6.
Hänel KH, Cornelissen C, Lüscher B, Baron JM. Cytokines and the skin barrier. Int J Mol Sci. 2013;14:6720–45.
Lee HJ, Kim M. Skin barrier function and the microbiome. Int J Mol Sci. 2022;23:13071.
Paller AS, Kong HH, Seed P, Naik S, Scharschmidt TC, Gallo RL, et al. The microbiome in patients with atopic dermatitis. J Allergy Clin Immunol. 2019;143:26–35.
Kobayashi T, Glatz M, Horiuchi K, Kawasaki H, Akiyama H, Kaplan DH, et al. Dysbiosis and staphylococcus aureus colonization drives inflammation in atopic dermatitis. Immunity. 2015;42:756–66.
Ali SM, Yosipovitch G. Skin ph: from basic science to basic skin care. Acta Derm Venereol. 2013;93:261–7.
Azimi E, Reddy VB, Lerner EA. Brief communication: Mrgprx2, atopic dermatitis and red man syndrome. Itch (Phila). 2017;2:e5.
Chiu IM, Heesters BA, Ghasemlou N, Von Hehn CA, Zhao F, Tran J, et al. Bacteria activate sensory neurons that modulate pain and inflammation. Nature. 2013;501:52–7.
Zebda R, Paller AS. Phosphodiesterase 4 inhibitors. J Am Acad Dermatol. 2018;78:S43–52.
Ständer S, Kwatra SG, Silverberg JI, Simpson EL, Thyssen JP, Yosipovitch G, et al. Early itch response with abrocitinib is associated with later efficacy outcomes in patients with moderate-to-severe atopic dermatitis: subgroup analysis of the randomized phase iii jade compare trial. Am J Clin Dermatol. 2022;24(1):97–107.
Han Y, Woo YR, Cho SH, Lee JD, Kim HS. Itch and janus kinase inhibitors. Acta Derm Venereol. 2023;103:adv00869.
Lin CM, Cooles FA, Isaacs JD. Basic mechanisms of jak inhibition. Mediterr J Rheumatol. 2020;31:100–4.
Simpson EL, Bieber T, Guttman-Yassky E, Beck LA, Blauvelt A, Cork MJ, et al. Two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N Engl J Med. 2016;375:2335–48.
Acknowledgements
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Medical Writing, Editorial, and Other Assistance
Editorial and medical writing support, under the guidance of the authors, was provided by Karis Moxley, PhD, and Lisa M. Klumpp Callan, PhD, at ApotheCom, San Francisco, CA, USA, and was funded by Pfizer Inc., New York, NY, USA, in accordance with the Good Publication Practice (GPP 2022) guidelines (Ann Intern Med. 2022; 10.7326/M22-1460).
Funding
The development of this manuscript was sponsored by Pfizer Inc. Publication costs were sponsored by Pfizer Inc.
Author information
Authors and Affiliations
Contributions
Sonja Ständer, Thomas Luger, Brian Kim, Ethan Lerner, Martin Metz, Roni Adiri, Juliana M. Canosa, Amy Cha, and Gil Yosipovitch: contributed equally to drafting and revising the article critically for important intellectual content and approved the final version to be published.
Corresponding author
Ethics declarations
Conflict of Interest
Roni Adiri, Juliana M. Canosa, and Amy Cha are employees and shareholders of Pfizer Inc. Ethan Lerner is a member of the Scientific Advisory Board for Escient Pharmaceuticals Inc. Martin Metz has received honoraria as a speaker and/or advisor for Pfizer Inc., AbbVie, Amgen, AstraZeneca, argenx, Bayer, Beiersdorf, Celldex, Escient, Galderma, GSK, Jasper, Novartis, Pharvaris, Sanofi-Aventis, Teva, ThirdHarmonicBio, and Vifor. Sonja Ständer was a speaker, consultant, and/or investigator and/or has received research funding from Pfizer Inc., AbbVie, Almirall, Beiersdorf, BMS, Clexio, Eli Lilly and Company, FomF, Galderma, German Research Foundation, Integrity CE, Kiniksa, LEO Pharma, L'Oréal, MEDahead, Moroscience, NACCME, Novartis, Omnicuris, P.G. Unna Academy, Sanofi, TouchIME, UCB, Vifor, and WebMD. Gil Yosipovitch has been a consultant and an advisor for Pfizer Inc., Arcutis, Bellus, Eli Lilly, Galderma, Kiniksa, LEO Pharma, Novartis, Pierre Fabre, Sanofi-Regeneron, Trevi Therapeutics, and a principal investigator for Pfizer Inc., Eli Lilly, Escient, Galderma, Kiniksa, LEO Pharma, Novartis, Pierre Fabre, and Sanofi-Regeneron. Thomas Luger has participated on advisory boards for Pfizer Inc., AbbVie, Amgen, Argenx, Celgene, Ceres Pharma, Eli Lilly, Galderma, Janssen, La Roche-Posay, LEO Pharma, Menlo Therapeutics, Mylan/Meda, Novartis, Pierre Fabre, PIQUR Therapeutics, Sandoz, Sanofi, and Symrise; acted as an investigator for Pfizer Inc., AbbVie, Celgene, Eli Lilly, Janssen, LEO Pharma, Menlo Therapeutics, Novartis, and Sandoz; participated as a speaker for Pfizer Inc., AbbVie, Galderma, Janssen, La Roche-Posay, Merck Sharp & Dohme, Mylan, Novartis, and Sanofi; and has received funding from Pfizer Inc., AbbVie, Celgene, Jansen-Cilag, Merck Sharp & Dohme, Mylan/Meda, Novartis, and Wolfe Laboratories. Brian Kim is a consultant and advisor for Pfizer Inc., AbbVie, Boehringer Ingelheim, Cara Therapeutics, Kiniksa, Menlo Therapeutics, and Sanofi-Regeneron; has received research grants from Cara Therapeutics, Celgene, and LEO Pharma; and is founder and stockholder in Nuogen Pharma. Brian Kim is currently affiliated with the Mark Lebwohl Center for Neuroinflammation and Sensation, Icahn School of Medicine at Mount Sinai, New York, NY, United States.
Ethical Approval
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Ständer, S., Luger, T., Kim, B. et al. Cutaneous Components Leading to Pruritus, Pain, and Neurosensitivity in Atopic Dermatitis: A Narrative Review. Dermatol Ther (Heidelb) 14, 45–57 (2024). https://doi.org/10.1007/s13555-023-01081-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13555-023-01081-0