
Abstract
Eosinophilic annular erythema (EAE) is a rare condition with a chronic relapsing and remitting course, characterized by the presence of annular or polycyclic erythematous and plaque lesions and prominent tissue eosinophilia on histopathology. There is an ongoing discussion on whether EAE is a subset of Wells syndrome (eosinophilic cellulitis) or a separate entity. To date, few cases of EAE have been reported in the literature; of these, about 40 cases were in adults and fewer than ten cases were in children. Given the rarity of this condition, there are no clear recommendations for its management. Systemic corticosteroids and antimalarials are the most commonly used medications used to treat EAE, but many cases have been reported in the literature that are resistant to treatment with these medications. Here, we present a 65-year-old female with EAE refractory to numerous systemic therapies (corticosteroids, hydroxychloroquine, dapsone, doxycycline, methotrexate) who showed a good response to mepolizumab, a humanized monoclonal antibody that blocks interleukin-5. To the best of our knowledge, this is the first reported case of mepolizumab therapy in a patient with EAE. We also review other treatment strategies that have been used to manage this condition to date. Targeting cytokines crucial for the functioning of eosinophils may be a novel direction in the management of EAE, but prospective, double-blinded and placebo-controlled studies are needed to provide further evidence.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Eosinophilic annular erythema is a rare, pruritic disease characterized by a chronic course and frequent resistance to treatment. |
There is a need for effective and safe therapeutic options for the management of eosinophilic annular erythema. |
Targeting cytokines crucial for the functioning of eosinophils, mainly interleukin-5, may be a novel direction in the treatment of eosinophilic annular erythema, but prospective, double-blinded and placebo-controlled studies are needed to provide further evidence. |
Introduction
Eosinophilic annular erythema (EAE) is a rare condition with a chronic relapsing and remitting course, characterized by the presence of annular or polycyclic erythematous and plaque lesions with a tendency to centrifugal spreading and prominent tissue eosinophilia on histopathology [1,2,3,4,5,6,7,8,9,10,11,12,13,14]. Skin lesions predominantly develop on the trunk and proximal aspects of the extremities. The condition was originally believed to be closely related to annular erythema of infancy (AEI). However, AEI and EAE are currently considered to be separate entities, with the former developing mainly in infants under the age of 1 year and spontaneously resolving, and the latter affecting predominantly adults [3].
A search of the literature revealed that only about 40 cases of EAE in adults and fewer than 10 cases in children have been reported to date. Among adult patients, a slight female preponderance has been noted (female:male ratio 1.14:1), and age at onset varies from 20 to 85 years [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17].
The aetiology of EAE remains unknown. Some authors consider this condition to be a result of a hypersensitivity reaction to an unidentified antigen [1, 4, 5]. EAE has been reported in association with autoimmune disorders, borreliosis, Helicobacter pylori infection, chronic hepatitis C, diabetes, and kidney disease [1, 4, 6, 7]. There are also a few reports of EAE developing in association with internal malignancy, such as thymoma [2], metastatic prostate adenocarcinoma [8], breast cancer, cervical cancer [9] and renal cancer [5]. In the reported case of EAE associated with thymoma, the lesions disappeared following thymectomy [2].
Given the rarity of this condition, there are no clear recommendations for its management. Here we report a patient with EAE refractory to numerous systemic therapies, who ultimately showed a good response to mepolizumab, a humanized monoclonal antibody that blocks interleukin (IL)-5. To the best of our knowledge, this is the first report of treating a patient with EAE with mepolizumab.
Informed consent was obtained from the patient for publication of the article, including the publication of clinical photographs.
Case report
A 65-year-old woman was referred to the Department of Dermatology for evaluation of widespread, pruritic annular erythematous lesions of 2-month duration. This was the third episode of such annular eruption in this patient, with the first one observed at the age of 29 years. In the previous two episodes, the lesions had resolved spontaneously over months to years, but in the third episode the lesions were resistant to treatment despite application of a wide range of therapies.
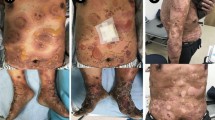
On admission, the physical examination revealed annular erythematous plaques with central hyperpigmentation located predominantly on the extremities (Fig. 1a, b). Test results from complete blood cell count and routine biochemistry tests (C-reactive protein, renal and liver function tests) were normal. Thyroid-stimulating hormone level was elevated and anti-thyroid peroxidase antibodies were present. Further extended laboratory tests (antinuclear antibodies, including SS-A and SS-B, anti-neutrophil cytoplasmic antibodies, screening antibodies for syphilis, hepatitis B and C, human immunodeficiency virus 1/2, Helicobacter pylori, Borrelia burgdorferi and pemphigus/pemphigoid) and radiographic studies (chest X-ray, computed tomography of the abdomen and pelvis and ultrasound of the peripheral lymph nodes) did not reveal any significant abnormalities. Multiple biopsies had been taken over the years. Recent histopathologic examinations found a normal epidermis and dense perivascular infiltration in the dermis that was composed of lymphocytes, histiocytes and eosinophils (Fig. 2a, b). There were no findings of either mucin deposits to suggest lupus erythematosus or “flame figures” typical of eosinophilic cellulitis (Wells syndrome). Direct immunofluorescence was negative for lupus erythematosus and autoimmune bullous diseases.

Clinical presentation of the lesions. a, b Sharply demarcated erythematous annular plaques with central hyperpigmentation, located on the dorsal aspects on hands (a) and lower legs (b). c, d Significant flattening of the borders of the lesions after first subcutaneous injection of mepolizumab 100 mg. e, f Complete resolution of the lesions on the upper limbs (e) and residual erythematous plaques on the lateral aspects of lower legs (f) 1 month after the third dose of mepolizumab

Histopathology of the erythematous border of the plaque. a Unchanged epidermis and dense perivascular infiltration in the dermis (×40, hematoxylin and eosin [H&E]). b Perivascular infiltration composed of numerous eosinophils, lymphocytes and histiocytes; “flame figures” were not found (×200, H&E)
On initial examination, several entities were taken into consideration in the differential diagnosis, including erythema annulare centrifugum, Wells syndrome, granuloma annulare, subacute cutaneous lupus erythematosus, erythema migrans, erythema gyratum repens and urticarial phase of bullous pemphigoid. However, based on the clinical presentation, laboratory findings (normal blood cell count without eosinophilia; negative antinuclear antibodies and negative serology for Borrelia burgdorferi, hepatitis B and C and screening antibodies for pemphigus/pemphigoid) and the histopathologic images (dense perivascular infiltration with abundant eosinophils and no “flame figures” or mucin deposits), these conditions were excluded and the diagnosis of EAE was made.
Treatment with oral prednisone (40 mg/day) was started, but the skin lesions continued to progress and the patient complained about gaining weight, leading to this therapy being discontinued after 1 month. Hydroxychloroquine (200 mg twice daily) was introduced and continued for 2 months, but this treatment did not lead to any improvement. Subsequent treatments with doxycycline (100 mg/day for 8 weeks) or methotrexate (10 mg/week for 6 weeks) were also not effective. Treatment with dapsone (100 mg/day) had to be discontinued after 1 month due to side effects (dyspnoea and haemolytic anaemia). While reviewing the literature, we came across a single case report of Wells syndrome that subsided completely following treatment with mepolizumab for eosinophilic granulomatosis with polyangiitis (EGPA; formerly referred to as Churg–Strauss syndrome) [18]. Mepolizumab is an anti-IL-5 monoclonal antibody that has proved to be effective in eosinophilic conditions, including asthma with eosinophilic phenotype, EGPA, chronic eosinophilic leukaemia and hypereosinophilic syndrome. Given the ineffectiveness of the standard treatment options to manage our patient’s condition, the presence of dense eosinophilic infiltrates on histopathologic images of the lesions and this report on the efficacy of mepolizumab in a presumably EAE-related entity, we considered initiating our patient on therapy with mepolizumab. Eventually, the patient was started on mepolizumab (100 mg subcutaneously every 4 weeks), as part of a 3-month program reimbursed by the public insurance by the National Health Found. Significant flattening of the lesions and alleviation of itching occurred after the first dose (Fig. 1c, d), and nearly complete resolution of skin changes was observed 1 month after the third injection of mepolizumab (Fig. 1e, f). In addition, the pruritus subsided completely. She did not report any side effects during therapy with mepolizumab. Given the patient’s good response, the treatment regimen was extended for another 3 months, but initiation of the treatment had to be postponed due to the outbreak of the coronavirus disease 2019 (COVID-19) pandemic. No relapse was observed within the 6-month period after the last (third) injection of mepolizumab, but some residual skin changes on the lower legs have persisted. At the time of writing this article, the patient has initiated the second cycle of mepolizumab treatment.
Discussion
The nosological position of EAE remains not fully elucidated. There is an ongoing discussion of whether EAE and Wells syndrome are related entities as both conditions share some similarities. It was initially believed that EAE may be differentiated from Wells syndrome by the absence of “flame figures” composed of eosinophilic major basic protein and degenerated collagen on histopathology and by a normal eosinophil count in peripheral blood (Table 1). Subsequently, however, several authors suggested that “flame figures” and peripheral blood eosinophilia may be present in well-developed and long-standing EAE and, therefore, the condition is currently considered by some to be a subset of Wells syndrome [4, 5].
There are no predefined diagnostic criteria for EAE, and the diagnosis should be based on: (1) presence of annular erythematous lesions characterized by a chronic relapsing and remitting course; (2) absence of blood eosinophilia (in the majority of cases); (3) histopathologic findings of dense perivascular infiltration composed of numerous eosinophils and absence of “flame figures” (except for long-lasting lesions); and (4) exclusion of other conditions (mainly Wells syndrome, urticaria, erythema annulare centrifugum, lupus erythematosus).
As the exact aetiology of EAE remains unknown, the treatment is mainly symptomatic and the mechanism of action is not fully elucidated. Systemic corticosteroids and antimalarials are the most commonly used treatment option. Antimalarials, when efficacious, lead to significant improvement within 2–4 weeks. Still, some authors reported observing a delayed response to chloroquine and decided to prolong treatment to avoid relapses [10]. Nevertheless, many cases resistant to treatment with systemic corticosteroids and antimalarials have been reported in the literature [1, 4, 5]. In one study, cyclosporine A was used mainly in combination with systemic corticosteroids, with several patients showing partial improvement [4]. A number of studies have reported some cases showing a good response to dapsone [11,12,13]. Howes et al. [14] presented a patient with EAE who had a striking response to indomethacin; however, the treatment had to be discontinued due to intolerable side effects. Nicotinamide (900 mg/day) was also successfully administered to one patient with EAE and autoimmune pancreatitis [6]. Methotrexate (10 mg/week) was found to be effective in a single case, but the treatment was discontinued after 2 weeks due to blood cell count abnormalities [7]. In the same patient, treatment with mycophenolate mofetil at a dose of 500 mg twice daily led to moderate improvement, but this treatment was stopped due to elevated liver enzyme levels, which were subsequently attributed to the onset of autoimmune hepatitis. There is also a single case report of an 8-year-old-boy who achieved complete resolution of EAE after eight sessions of narrow-band ultraviolet B phototherapy [15].
Eosinophils are considered to be the crucial cells in the development of EAE. The efficacy of antimalarials has been suggested to be associated to their ability to inhibit eosinophilotaxis and release proinflammatory cytokines [1]. Dapsone has been found to inhibit eosinophil peroxidase and block mast cell degranulation and activation. Data from recent case reports underscore the efficacy of targeting Th2-type cytokines in the treatment of EAE. Suplatast tosilate, an anti-allergic medication that suppresses the production of IgE and synthesis of IL-4 and IL-5, was administered to one patient with EAE, resulting in complete resolution of the lesions within 2 weeks [16]. Dupilumab, an IL-4 receptor alpha antagonist, was also successfully used in a 14-year-old African-American girl with EAE [17].
Mepolizumab is a humanized monoclonal antibody targeting IL-5, a cytokine that is crucial for the development and tissue migration of eosinophils. The medication is used in the treatment of severe eosinophilic asthma. It is also approved by the U.S. Food and Drug Administration for the treatment of EGPA. There has been one published report of a patient with EGPA coexisting with Wells syndrome, in whom treatment with mepolizumab resulted in complete resolution of the skin lesions [18]. To the best of our knowledge, mepolizumab has not been used previously to treat EAE. In the patient presented here, the initial dose of mepolizumab led to significant improvement of the skin condition and severity of pruritus. According to the patient, the effect was better than that of all previous treatment regimens. Skin lesions continued to subside after subsequent injections, and nearly complete remission was observed 1 month after the third dose of mepolizumab. Good response to an anti-IL-5 agent speaks for an important role of eosinophils in the pathogenesis of EAE and might set a new direction in the management of the condition.
Conclusion
Eosinophilic annular erythema is a pruritic disease with a chronic course and frequent resistance to treatment. There is a need for effective and safe therapeutic options for the condition. Targeting cytokines crucial for the functioning of eosinophils may be a novel direction in the treatment of EAE, but prospective, double-blinded and placebo-controlled studies are needed to provide further evidence.
References
Kahofer P, Grabmaier E, Aberer E. Treatment of eosinophilic annular erythema with chloroquine. Acta Derm Venereol. 2000;80:70–1.
Iga N, Otsuka A, Kaku Y, Miyachi Y, Kabashima K. Eosinophilic annular erythema limited on the palms and the soles and possibly associated with thymoma. J Eur Acad Dermatol Venereol. 2016;30:1213–4.
Peterson AO, Jarrat M. Annular erythema of infancy. Arch Dermatol. 1981;117:145–8.
El-Khalawany M, Al-Mutairi N, Sultan M, Shaaban D. Eosinophilic annular erythema is a peculiar subtype in the spectrum of Wells syndrome: a multicentre long-term follow-up study. J Eur Acad Dermatol Venereol. 2013;27:973–9.
Rongioletti F, Fausti V, Kempf W, Rebora A, Parodi A. Eosinophilic annular erythema: an expression of the clinical and pathological polymorphism of wells syndrome. J Am Acad Dermatol. 2011;65:e135–e137137.
Ogawa K, Fukumoto T, Yoshida M, et al. Eosinophilic annular erythema in a patient with autoimmune pancreatitis: nicotinamide therapy may be beneficial for achieving remission. J Dermatol. 2016;43:1380–1.
Awosika O, Totoraitis K, Eleryan M, Rengifo-Pardo M, Ehrlich A. A case of eosinophilic annular erythema as a presenting sign for autoimmune hepatitis. JAAD Case Rep. 2017;4:84–6.
Gonzalez-Lopez MA, Lopez-Escobar M, Fernandez-Llaca H, Gonzalez-Vela MC, Lopez-Brea M. Eosinophilic annular erythema in a patient with metastatic prostate adenocarcinoma. Int J Dermatol. 2015;54:e80–e8282.
Nakazato S, Fujita Y, Shinkuma S, Nomura T, Shimizu H. Eosinophilic annular erythema is clinically characterized by central pigmentation reflecting basal melanosis: a clinicopathological study of 10 cases. J Eur Acad Dermatol Venereol. 2017;31:1916–23.
Ljubojević Hadzavdić S, Bartolić L, Bradamante M. Prolonged treatment of eosinophilic erythema annulare with chloroquine. Acta Dermatovenereol Croat. 2018;26:262–3.
Manriquez J, Berroeta-Mauriziano D, Andino-Navarrete R, Vera-Kellet C. Eosinophilic annular erythema: complete clinical response with dapsone. Int J Dermatol. 2015;54:e96–e9898.
Wallis L, Gilson RC, Gilson RT. Dapsone for recalcitrant eosinophilic annular erythema: a case report and literature review. Dermatol Ther (Heidelb). 2018;8:157–63.
Alharbi R, Peric J, Wollf H, Wollenberg A. Dapsone treatment for eosinophilic annular erythema. J Eur Acad Dermatol Venereol. 2017;31:e153–e154154.
Howes R, Girgis L, Kossard S. Eosinophilic annular erythema: a subset of Wells’ syndrome or a distinct entity? Australas J Dermatol. 2008;49:159–63.
Thomas L, Fatah S, Nagarajan S, Natarajan S. Eosinophilic annular erythema: successful response to ultraviolet B therapy. Clin Exp Dermatol. 2015;40:883–6.
Kanameishi S, Goto K, Ogawa M, Fukumoto T, Tanabe H. Efficacy of suplatast tosilate in a case of recurrent eosinophilic annular erythema. Int J Dermatol. 2018;35:e255–e256256.
Gordon SC, Robinson SN, Abudu M, et al. Eosinophilic annular erythema treated with dupilumab. Pediatr Dermatol. 2018;35:e255–e256256.
Herout S, Bauer WM, Schuster C, Stingl G. Eosinophilic cellulitis (Wells syndrome) successfully treated with mepolizumab. JAAD Case Rep. 2018;4:548–50.
Acknowledgements
We would like to thank the patient for her cooperation.
Funding
Treatment with mepolizumab was initiated as part of 3-month program reimbursed by the National Health Found. No Rapid Service Fee was received by the journal for the publication of this article.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Authorship Contributions
Magdalena Żychowska and Klaudia Tutka contributed equally to the article.
Disclosures
Klaudia Tutka and Magdalena Żychowska have nothing to disclose. Adam Reich is a member of the journal’s Editorial Board.
Compliance with Ethics Guidelines
Informed consent was obtained from the patient for publication of the article, including publication of clinical photographs.
Data Availability
Data sharing is not applicable to this article as no data sets were generated or analyzed during the current study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Digital Features
To view digital features for this article go to https://doi.org/10.6084/m9.figshare.12458429.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Żychowska, M., Tutka, K. & Reich, A. Mepolizumab Therapy for Recalcitrant Eosinophilic Annular Erythema in an Adult: A Case Report and Review of Treatment Options. Dermatol Ther (Heidelb) 10, 893–899 (2020). https://doi.org/10.1007/s13555-020-00412-9
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13555-020-00412-9