
Abstract
Purpose
This study was aimed to identify biomarker associated with membranous nephropathy (MN) progression by integration of expression profiles and competitive endogenous RNA (ceRNA) network analysis.
Methods
The gene (GSE108113) and microRNAs (miRNAs) expression profiles (GSE64306) were downloaded to identify the differentially expressed mRNAs, miRNAs and long non-coding RNAs (lncRNAs) between MN and control groups. The functions and pathways enriched by the differentially expressed mRNAs were analyzed. The mRNA-lncRNA co-expression network was constructed followed by and the ceRNA network construction.
Results
Total 264 upregulated and 196 downregulated differentially expressed mRNAs, 79 upregulated and 4 downregulated lncRNAs, as well as 115 upregulated and 93 downregulated miRNAs were obtained between MN and control groups. After analysis, the differential mRNAs were significantly involved in multiple immune-related processes, and cell proliferation, apoptosis and differentiation processes, as well as pathways of taste transduction and lysosome. Finally, a ceRNA network consisting of 4 mRNAs (EPB41L5, FAM43A, PRKG1 and TTC14), 3 lncRNAs (LINC00052, LINC00641 and N4BP2L2-IT2) and 5 miRNAs (hsa-miR-145-5p, hsa-miR-3605-5p, hsa-miR-148a-3p, hsa-miR-497-5p and hsa-miR-148b-3p) was constructed.
Conclusion
Our study indicated dysregulation of immune- and apoptosis-associated functions and taste transduction and lysosome pathways may play important roles in MN progression. Deregulated ceRNAs, such as LINC00052-hsa-miR-145-5p-EPB41L5, LINC00052-hsa-miR-148a-3p-FAM43A and LINC00641-hsa-497-5p-PRKG1, may be associated with MN development.
Similar content being viewed by others
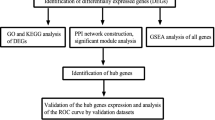


1 Introduction
Membranous nephropathy (MN) is a unique glomerular lesion characterized by the uniform thickening of the basement membrane because of the sub-epithelial deposition of complement components and immune complexes, as well as the synthesis of new basement membrane [1, 2]. In adults, MN often results in nephrotic syndrome, which eventually progresses to end-stage renal failure in some patients [3, 4]. There is presently no specific treatment for MN [5]. Therefore, it is urgent to understand the pathogenesis of MN so as to identify new factors that may facilitate the development of new treatments for this complex disease.
The improvement of high-throughput sequencing techniques has identified more than 100,000 RNAs, which are far from than previous estimated protein-coding genes (about 20,000) [6, 7]. The non-coding RNAs, such as microRNAs (miRNAs) and long non-coding RNAs (lncRNAs), are found to be involved in diverse biological processes and diseases [8, 9]. Microarray analysis has been used in the field of experimental nephrology to investigate human diseases [10,11,12]. However, similar study of MN has been rarely reported [13]. Recent studies have shown that lncRNAs can regulate the expression of other transcripts by acting as a miRNA sponge, thereby competing with endogenous RNA [14, 15]. Xiao et al. [16] discovered that the lncRNA MALAT1 acted as a competing endogenous RNA (ceRNA) in clear cell kidney cancer via sponging miR-200 s. Studies have found that the integration of gene expression profiles and ceRNA networks can identify risk biomarkers [17]. Nevertheless, this integration analysis has not been applied in MN.
In this study, we intended to identify biomarker associated with MN progression by integration of the gene expression profiles and ceRNA network analysis. Thus, the gene and miRNA expression profiles were downloaded to identify the differential genes between MN and control groups, and then construct the ceRNA network. The biomarkers discovered could be used as diagnostic and therapeutic targets for MN.
2 Materials and Methods
2.1 Data Downloads and Gene Re-annotation
The gene expression profile data GSE108113 (GPL19983) for MN were downloaded from the GEO database, which included gene expression data of 87 MN samples and 41 control samples. Additionally, the miRNA expression profiles GSE64306 (GPL19117) were also downloaded, which included 4 MN and six control samples. The mRNAs and lncRNAs were annotated according to the gtf gene annotation file in the GENCODE database [18]. The gene whose annotation information is “protein coding” is reserved as a mRNA, and with annotation of “processed transcript”, “antisense”, “sense overlapping”, “lincRNA”, “3 prime overlapping ncrna”, “sense intronic”, or “non-coding” was reserved as a lncRNA. The probes that did not match a gene symbol were deleted. The average value of the probes was used to compute the final expression value of genes when numerous probes corresponded to the same gene symbol.
As a validation dataset, the GSE133288 dataset was downloaded from the GEO database, which included gene expression data from 48 MN samples and five control samples. The platform of this dataset was GPL19983.
2.2 Differential Expression Analysis
For the datasets of genes and miRNAs, the limma package was used to perform differential expression analysis for MN vs. control [19], and p values of all genes were obtained after statistical test. The Benjamini and Hochberg (BH) approach was used to correct multiple tests. The threshold values for differentially expressed lncRNAs (DE-lncRNAs) and mRNAs (DE-mRNAs) were |log fold change (FC)|≥ 1 and adjusted p value ≤ 0.05 and for differentially expressed miRNAs (DE-miRNAs) were p value ≤ 0.05 and |log FC|≥ 1.
2.3 Gene Ontology (GO) Functions and the Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Enrichment Analysis
The DAVID version 6.7 (https://david-d.ncifcrf.gov/) was used to analyze the GO functions of DE-mRNAs enrichment [20]. GOplot [21] was used to visualize the results. The Gene set enrichment analysis (GSEA) [22] version 3.0 was used to carry out the KEGG [23] pathway enrichment analysis.
2.4 The mRNA–lncRNA Co-expression Network
The corr.test in R psych [24] (parameter ci = F, adjust = “BH”) was used to calculate the Pearson correlation coefficient (PCC) between DE-mRNAs and DE-lncRNAs. The multiple test correction was performed with BH method. The network was visualized through Cytoscape 3.7.0 [25] (http://www.cytoscape.org/).
2.5 The miRNA Prediction and ceRNA Network Analysis
Based on the starbase [26] (version 3.0, http://starbase.sysu.edu.cn/), the lncRNAs in the above co-expression relations (lncRNA–mRNA) were subjected to lncRNA–miRNA relation prediction. For the mRNAs in the lncRNA–mRNA co-expression relations, the tool mirwalk [27] (version 3.0) was used for miRNA-mRNA prediction by combining the Targetscan, Mirdb, and Mirtarbase databases. The mRNA–miRNA relation pairs appeared in at least two databases were selected. Then, from the obtained co-expression relation pairs, the relation pairs that were negatively regulated by the same DE-miRNAs were screened. The ceRNA network was established and visualized using Cytoscape. To investigate the function of ceRNA, we analyzed the function of mRNA in this network using the GeneCards database [28].
2.6 Validation of DE-lncRNAs and DE-mRNAs
The GSE133288 dataset was adopted for validating the expression of candidate mRNAs and lncRNAs. The transcriptome profiles in the GSE133288 (Platform GPL19983) dataset included 48 MN samples and five control samples. p < 0.05 was considered to the statistical significant level.
3 Results
3.1 Gene Differential Expression Analysis
There were 264 upregulated and 196 downregulated DE-mRNAs, 79 upregulated and four downregulated DE-lncRNAs, as well as 115 upregulated and 93 downregulated DE-miRNAs were obtained between MN and control groups. The volcano plot and hierarchical clustering heatmap are shown in Fig. 1.

Analysis of differentially expressed long non-coding RNAs (lncRNAs), microRNAs (miRNAs) and mRNAs in membranous nephropathy (MN) from GEO databases. Volcano plots of differentially expressed mRNAs (A), lncRNAs (C), and miRNAs (E). The Y-axis represents the negative log (base 10) of the FDR, and the X-axis represents the log (base 2) of FC. Red dots represent upregulation while green dots represent downregulation. Heatmap plots of the top 10 differentially expressed mRNAs (B), lncRNAs (D), and miRNAs (F) in descending order by log FC. Differential expressed genes are represented in rows, and samples are represented in columns
3.2 GO Enrichment Analysis
DE-mRNAs were significantly enriched in 131 biological processes (BP), 10 cell components (CC), and eight molecular functions (MF). Some significant enrichment results are shown in Fig. 2. In terms of CC, the DE-mRNAs mainly functioned on the cell membrane and were closely related to adherens junction, intrinsic to membrane and cell-substrate adherens junction. From the point of MF, the DE-mRNAs were significantly associated with taste receptor activity, protein hetero-dimerization activity, cation binding, and metal ion binding. From the perspective of BP, the DE-mRNAs involved in multiple immune-related processes, and cell proliferation, differentiation and apoptosis processes.

Gene ontology (GO) functions enriched by differentially expressed mRNAs. The top five bubble plot of GO terms. X-axis represents the Z-score and Y-axis represents the negative log (adjusted p value). The area of the bubble positively correlates with the gene numbers in the indicated term. The green represents the GO-biological process terms, the pink represents the GO-cellular component terms and yellow represents the GO-molecular function
3.3 KEGG Pathway Analysis
According to the GSEA analysis, a total of 147 pathways with significant enrichment were identified, among which, 34 were upregulated (normalized enrichment score (NES) > 0) and 113 were downregulated (NES < 0). Some significant results are shown in Fig. 3A, B. Taste transduction had the maximum NES among the upregulated pathways (Fig. 3C). Additionally, CGMP-PKG signaling pathway, TNF signaling pathway, IL17 signaling pathway, cell adhesion molecules, and adherens junction also had high NES. Among the significant downregulated pathways, lysosome had the maximum NES (Fig. 3B).

KEGG pathway enrichment analysis of differentially expressed mRNAs. The KEGG pathway enrichment of differentially expressed mRNAs was performed by Gene set enrichment analysis. A The dotplot of partially enriched pathways. The color intensity of the nodes indicates KEGG pathways enrichment degree. Horizontal axis indicates the gene ratio as the proportion of differential genes in the whole gene set. The size represents the number counts in a certain pathway. B The joyplot of the up- and downregulated pathways. Horizontal axis represents p value. C, D Genes involved in TASTE TRANSDUCTION pathway and LYSOSOME pathway. Running enrichment score was positive for most genes in TASTE TRANSDUCTION pathway and negative for most genes in LYSOSOME pathway
3.4 Co-expression Network
The highly co-expressed mRNA–lncRNA pairs with r ≥ 0.7 and adjusted p < 0.05 were selected out based on the correlation between mRNA and lncRNA expression values. A co-expression network was constructed, which included 150 nodes (17 lncRNAs and 133 mRNAs) and 440 edges (Fig. 4). Some lncRNAs interacted with multiple mRNAs, such as FRY-AS1, TARID, LINC01031, LINC02149, LINC01116, ISX-AS1, CHRM3-AS2, DNM1P35, LINC00052, and so on in the network.

LncRNA–mRNA co-expression network constructed using differentially expressed lncRNAs and mRNAs. Pearson's correlation coefficients (PCC) between differentially expressed lncRNAs and mRNAs were calculated using psych package in R and the co-expressed lncRNA–mRNA pairs with adjusted p value < 0.05 and r ≥ 0.7 were visualized by Cytoscape. Red: mRNA, yellow: lncRNA
3.5 miRNA Prediction and ceRNA Network Construction
On the basis of lncRNA–mRNA, 558 miRNA–mRNA pairs that appeared in at least two databases were predicted (Fig. 5A). miRNA–lncRNA and miRNA–mRNA were then integrated and filtered by DE-miRNAs, and the ceRNA network was constructed, as shown in Fig. 5B. The network consisted of four mRNAs (erythrocyte membrane protein band 4.1 like 5 (EPB41L5), family with sequence similarity 43 member A (FAM43A), protein kinase CGMP-dependent 1 (PRKG1) and tetratricopeptide repeat domain 14 (TTC14)), 3 lncRNAs (LINC00052, LINC00641 and N4BP2L2-IT2) and 5 miRNAs (hsa-miR-145-5p, hsa-miR-148a-3p, hsa-miR-148b-3p, hsa-miR-3605-5p and hsa-miR-497-5p). As shown in the ceRNA network, LINC00052 may act as the ceRNA of hsa-miR-145-5p, hsa-miR-148b-3p and hsa-miR-148a-3p to increase the expression of FAM43A and EPB41L5 in MN. N4BP2L2-IT2 may play a role in MN by down-regulating hsa-miR-3605-5p to increase the expression of TTC14. LINC00641 may target hsa-miR-497-5p to regulate the expression of PRKG1.

CeRNA network construction. A Venn diagram shows the number of predicted miRNA‐mRNA interactions from TargetScan, MirDB, MirTarBase. B ceRNA network. Pink nodes were mRNAs, yellow nodes were lncRNAs, and blue nodes were miRNAs
3.6 Validation of DE-mRNAs and DE-lncRNAs
To verify our analysis, the expression levels of EPB41L5, FAM43A, PRKG, TTC14, LINC00052, LINC00641 and N4BP2L2-IT2 were assessed in 48 MN samples and five control samples from GSE133288. Consistent with our findings, the average expression levels of EPB41L5, FAM43A, PRKG, TTC14 and N4BP2L2-IT2 were significantly higher in MN samples than those in control samples in the GSE133288 dataset (p < 0.05) (Fig. 6). The results proved the reliability of our analysis.

The expression levels of EPB41L5, FAM43A, PRKG, TTC14 and N4BP2L2-IT2 in the GSE133288 validation dataset. p < 0.05 was regarded as the statistically significant level
4 Discussion
In this study, we identified hundreds of differentially expressed genes. After analysis, the DE-mRNAs were significantly involved in multiple immune-related processes, and cell proliferation, differentiation and apoptosis processes, as well as pathways of taste transduction and lysosome. Finally, a ceRNA network consisted of 4 mRNAs (EPB41L5, FAM43A, PRKG1 and TTC14), 3 lncRNAs (LINC00052, LINC00641 and N4BP2L2-IT2) and 5 miRNAs (hsa-miR-145-5p, hsa-miR-148a-3p, hsa-miR-3605-5p, hsa-miR-148b-3p and hsa-miR-497-5p) was constructed.
It is well known that MN is an immune-mediated glomerular disease [29]. The results of our study also showed that the DE-mRNAs, such as complement C3a receptor 1 (C3AR1), interleukin 1 receptor-associated kinase 3 (IRAK3) and complement C3b/C4b receptor 1 (CR1), were significantly enriched in immune-related GO functions, which may indicate the reliability of our results. Immune-mediated podocyte injuries are critical in the progression of proteinuria in MN [30]. It has been reported that podocytes apoptosis is a critical process during the MN development [31]. In the present study, the DE-mRNAs were also involved in apoptosis related functions, such as induction of apoptosis by intracellular signals [BCL2-associated X, BCL3 transcription coactivator (BCL3), tumor protein P53 (TP53) and apoptosis regulator (BAX)]. We speculated that BAX, TP53 and BCL3 may serve as targets to inhibit the podocytes apoptosis in the treatment of MN.
Taste is a crucial sense for determining the nutritional value of food and detecting and avoiding the absorption of potentially hazardous chemicals [32, 33]. Numerous diseases are found to be associated with taste dysfunction [34]. It has been reported that certain foods appear to taste ‘different’ for patients with chronic kidney disease. Additionally, the degree of taste disturbance in chronic kidney disease appears to be related to the severity of kidney dysfunction [35]. The present study revealed that pathway of taste transduction was significantly upregulated. Importantly, this pathway is also abnormal in diabetic nephropathy, renal failure, and various cancers [36,37,38]. Thus, dysregulation of taste transduction may play a key role in MN progression.
Moreover, among the significant downregulated pathways, lysosome had the maximum NES. The lysosome is a single-membrane acidic organelle, which acts as a cellular ‘digestive apparatus’ to degrade the materials delivered from inside or outside [39]. Lysosomal rupture can result in content leakage from the lysosomal lumen into the cytosol. Diverse substances, such as cell death effectors bacterial, mineral crystals and lipids, can damage lysosomal membranes in vivo, thereby causing pathologies, including the development of hyperuricemic nephropathy [40]. We speculated that lysosomal rupture may be a potential even leading to MN.
LINC00052 was involved in three ceRNA pathway, such as LINC00052-hsa-miR-145-5p-EPB41L5 and LINC00052-hsa-miR-148a-3p-FAM43A in the ceRNA network. Hsa-miR-145-5p is reported to be dysregulated in renal cell carcinoma [41]. Study has suggested that EPB41L5 expression is restricted to the lung, brain, and kidney. In the kidney, EPB41L5 is particularly strongly expressed in the embryonic glomeruli. Loss of EPB41L5 can result in renal failure and nephrotic syndrome [42]. A recent study demonstrated that miR-148a-3p was upregulated in renal cell carcinomas compared with normal renal parenchyma. The role of FAM43A in kidney disease has not been reported to our best knowledge. Furthermore, LINC00641 act as a ceRNA by sponging hsa-497-5p to regulate PRKG1. A recent study demonstrated that miR-497-5p can inhibit cell proliferation, while promoted apoptosis in clear cell renal cell carcinoma. Given the roles of hsa-miR-145-5p, hsa-497-5p, hsa-miR-148a-3p and EPB41L5 in kidney diseases, we speculated that aberrations in these genes may deregulate ceRNAs LINC00052-hsa-miR-145-5p-EPB41L5, LINC00052-hsa-miR-148a-3p-FAM43A and LINC00641-hsa-497-5p-PRKG1 and thus lead to MN.
This study has several limitations. First, the samples size was small. Second, mRNA, lncRNA and miRNA data are not from the same data set. Additionally, the results were not verified through in vivo and in vitro experiments. Therefore, further animal or clinical experiments with more samples are needed to validate our results.
5 Conclusion
Dysregulation of immune and apoptosis-associated function and taste transduction and lysosome pathways may play important roles in MN progression. Deregulated ceRNAs, such as LINC00052-hsa-miR-145-5p-EPB41L5, LINC00052-hsa-miR-148a-3p-FAM43A and LINC00641-hsa-497-5p-PRKG1, may be associated with MN development.
References
Balafa O, Kalaitzidis R, Liapis G, Xiromeriti S, Zarzoulas F, Baltatzis G, Elisaf M (2015) Crescentic glomerulonephritis and membranous nephropathy: a rare coexistence. Int Urol Nephrol 47(8):1373–1377
Couser WG (2017) Primary membranous nephropathy. Clin J Am Soc Nephrol 12(6):983–997
Ponticelli C, Passerini P (2010) Can prognostic factors assist therapeutic decisions in idiopathic membranous nephropathy. J Nephrol 23(2):156–163
Cybulsky AV, Quigg RJ, Salant DJ (2005) Experimental membranous nephropathy redux. Am J Physiol-Renal Physiol 289(4):F660–F671
Chen W, Lin X, Huang J, Tan K, Chen Y, Peng W, Li W, Dai Y (2014) Integrated profiling of microRNA expression in membranous nephropathy using high-throughput sequencing technology. Int J Mol Med 33(1):25–34
Hon CC, Ramilowski JA, Harshbarger J, Bertin N, Rackham OJL, Gough J, Denisenko E, Schmeier S, Poulsen TM, Severin J (2017) An atlas of human long non-coding RNAs with accurate 5′ ends. Nature 543(7644):199–204
Iyer MK, Niknafs YS, Rohit M, Udit S, Anirban S, Yasuyuki H, Barrette TR, Prensner JR, Evans JR, Shuang Z (2015) The landscape of long noncoding RNAs in the human transcriptome. Nat Genet 47(3):199–208
Alessandro F, Irene B (2014) Long non-coding RNAs: new players in cell differentiation and development. Nat Rev Genet 15(3):7–21
Boen JR, Gevaert AB, De Keulenaer GW, Van Craenenbroeck EM, Segers VF (2019) The role of endothelial miRNAs in myocardial biology and disease. J Mol Cell Cardiol 138:75–87
Higgins JP, Shinghal R, Gill H, Reese JH, Terris M, Cohen RJ, Fero M, Pollack JR, Van de Rijn M, Brooks JD (2003) Gene expression patterns in renal cell carcinoma assessed by complementary DNA microarray. Am J Pathol 162(3):925–932
Perco P, Pleban C, Kainz A, Lukas A, Mayer B, Oberbauer R (2007) Gene expression and biomarkers in renal transplant ischemia reperfusion injury. Transpl Int 20(1):2–11
Susztak K, Böttinger E, Novetsky A, Liang D, Zhu Y, Ciccone E, Wu D, Dunn S, McCue P, Sharma K (2004) Molecular profiling of diabetic mouse kidney reveals novel genes linked to glomerular disease. Diabetes 53(3):784–794
Stanescu HC, Arcos-Burgos M, Medlar A, Bockenhauer D, Kottgen A, Dragomirescu L, Voinescu C, Patel N, Pearce K, Hubank M (2011) Risk HLA-DQA1 and PLA2R1 alleles in idiopathic membranous nephropathy. N Engl J Med 364(7):616–626
Ebert MS, Sharp PA (2010) Emerging roles for natural microRNA sponges. Curr Biol 20(19):R858–R861
Sen R, Ghosal S, Das S, Balti S, Chakrabarti J (2014) Competing endogenous RNA: the key to posttranscriptional regulation. Sci World J 2014:896206
Xiao H, Tang K, Liu P, Chen K, Hu J, Zeng J, Xiao W, Yu G, Yao W, Zhou H (2015) LncRNA MALAT1 functions as a competing endogenous RNA to regulate ZEB2 expression by sponging miR-200s in clear cell kidney carcinoma. Oncotarget 6(35):38005
Cao Y, Wang P, Ning S, Xiao W, Xiao B, Li X (2016) Identification of prognostic biomarkers in glioblastoma using a long non-coding RNA-mediated, competitive endogenous RNA network. Oncotarget 7(27):41737
Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S et al (2012) GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res 22(9):1760–1774
Bradizza CM, Stasiewicz PR, Paas ND (2006) Relapse to alcohol and drug use among individuals diagnosed with co-occurring mental health and substance use disorders: a review. Clin Psychol Rev 26(2):162–178
da Huang W, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4(1):44–57
Chiasson PM, Pace DE, Schlachta CM, Mamazza J, Poulin EC (2003) Minimally invasive surgery training in Canada: a survey of general surgery. Surg Endosc 17(3):371–377
Pruitt KD, Brown GR, Hiatt SM, Thibaud-Nissen F, Astashyn A, Ermolaeva O, Farrell CM, Hart J, Landrum MJ, McGarvey KM et al (2014) RefSeq: an update on mammalian reference sequences. Nucleic Acids Res 42(Database issue):D756–D763
Tikole S, Sankararamakrishnan R (2006) A survey of mRNA sequences with a non-AUG start codon in RefSeq database. J Biomol Struct Dyn 24(1):33–42
Jason M (2013) Psych issues. JEMS 38(3):14
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13(11):2498–2504
Li JH, Liu S, Zhou H, Qu LH, Yang JH (2014) starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res 42(Database issue):D92–D97
Sticht C, De La Torre C, Parveen A, Gretz N (2018) miRWalk: An online resource for prediction of microRNA binding sites. PLoS One 13(10):e0206239
Safran M, Dalah I, Alexander J, Rosen N, Iny Stein T, Shmoish M, Nativ N, Bahir I, Doniger T, Krug H et al (2010) GeneCards Version 3: the human gene integrator. Database J Biol Databases Curation 2010:baq020
Roccatello D, Sciascia S, Di Simone D, Solfietti L, Naretto C, Fenoglio R, Baldovino S, Menegatti E (2016) New insights into immune mechanisms underlying response to Rituximab in patients with membranous nephropathy: a prospective study and a review of the literature. Autoimmun Rev 15(6):529–538
Chen Z-H, Qin W-S, Zeng C-H, Zheng C-X, Hong Y-M, Lu Y-Z, Li L-S, Liu Z-H (2010) Triptolide reduces proteinuria in experimental membranous nephropathy and protects against C5b–9-induced podocyte injury in vitro. Kidney Int 77(11):974–988
Sha W-g, Shen L, Zhou L, Xu D-y, Lu G-y (2015) Down-regulation of miR-186 contributes to podocytes apoptosis in membranous nephropathy. Biomed Pharmacother 75:179–184
Hovan AJ, Williams PM, Stevenson-Moore P, Wahlin YB, Ohrn KE, Elting LS, Spijkervet FK, Brennan MT (2010) A systematic review of dysgeusia induced by cancer therapies. Support Care Cancer 18(8):1081–1087
Epstein JB, Barasch A (2010) Taste disorders in cancer patients: pathogenesis, and approach to assessment and management. Oral Oncol 46(2):77–81
Bromley SM, Doty RL (2015) Clinical disorders affecting taste: an update. In: Handbook of olfaction and gustation, pp 887–910
Armstrong JE, Laing DG, Wilkes FJ, Kainer G (2010) Smell and taste function in children with chronic kidney disease. Pediatr Nephrol 25(8):1497–1504
Zhou L, Huang W, Xu Y, Gao C, Zhang T, Guo M, Liu Y, Ding J, Qin L, Xu Z et al (2018) Sweet taste receptors mediated ROS-NLRP3 inflammasome signaling activation: implications for diabetic nephropathy. J Diabetes Res 2018:7078214
Vivas L, Godino A, Dalmasso C, Caeiro XE, Macchione AF, Cambiasso MJ (2014) Neurochemical Circuits Subserving Fluid Balance and Baroreflex: a role for serotonin, oxytocin, and gonadal steroids. In: De Luca LA Jr, Menani JV, Johnson AK (eds) Neurobiology of body fluid homeostasis: transduction and integration, Boca Raton
Liu S, Xu M, Zhu C, Zhao Q, Zhou F (2018) Taste receptor T1R1/T1R3 promotes the tumoricidal activity of hepatic CD49a(+) CD49b(-) natural killer cells. Eur J Immunol 48(12):2031–2041
Saftig P, Klumperman J (2009) Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat Rev Mol Cell Biol 10(9):623
Salminen A, Kaarniranta K, Kauppinen A (2012) Inflammaging: disturbed interplay between autophagy and inflammasomes. Aging 4(3):166
Liep J, Kilic E, Meyer HA, Busch J, Jung K, Rabien A (2016) Cooperative effect of miR-141-3p and miR-145-5p in the regulation of targets in clear cell renal cell carcinoma. PLoS One 11(6):e0157801
Schell C, Rogg M, Suhm M, Helmstädter M, Sellung D, Yasuda-Yamahara M, Kretz O, Küttner V, Suleiman H, Kollipara L (2017) The FERM protein EPB41L5 regulates actomyosin contractility and focal adhesion formation to maintain the kidney filtration barrier. Proc Natl Acad Sci 114(23):E4621–E4630
Acknowledgements
This research was supported by the Science and Technology Development Plan Project of Jilin Province (No. 20200201437JC and No. 20200201383JC), and the Special Project for Health Research of jilin Province (No. 2020SCZ37).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The author declares that they have no conflicts of interest.
Rights and permissions
About this article

Cite this article
Zhou, G., Jiang, N., Zhang, W. et al. Biomarker Identification in Membranous Nephropathy Using a Long Non-coding RNA-Mediated Competitive Endogenous RNA Network. Interdiscip Sci Comput Life Sci 13, 615–623 (2021). https://doi.org/10.1007/s12539-021-00466-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12539-021-00466-z